
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Мукополисахаридоз I типа (синдром Гурлер) у детей - причины, клиника
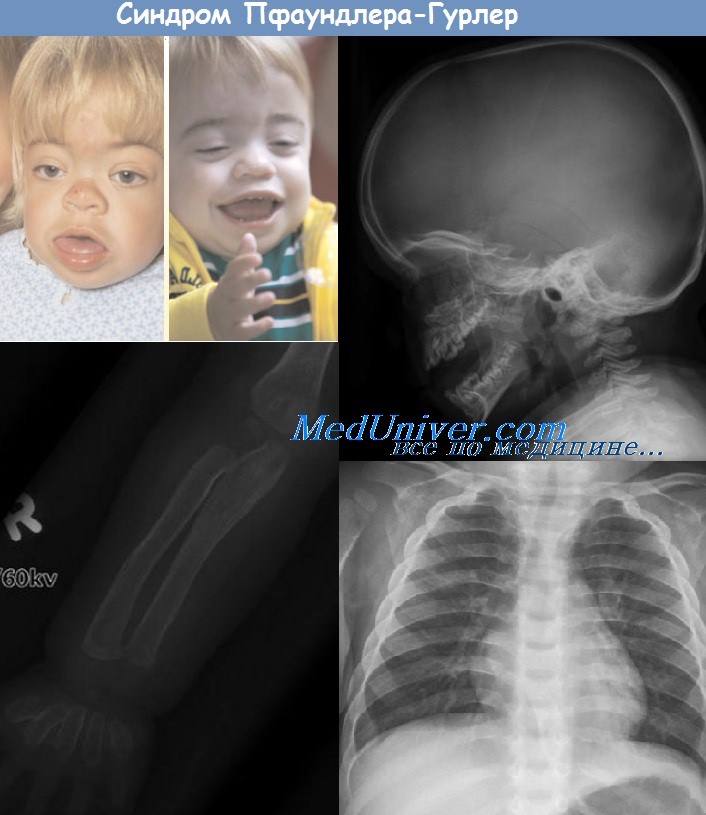
Заболевание характеризуется грубыми поражениями опорно-двигательного аппарата, выраженной умственной отсталостью, помутнением роговицы глаз, пороками сердечно-сосудистой системы, гепатоспленомегалией, прогрессирующим течением. У гетерозиготных носителей в моче обнаруживается повышенное содержание кислых ГАГ и их аномальных фракций (Tellek и соавт.).
Согласно исследованиям патоморфологов, мукополисахариды накапливаются во многих органах больного. Особенно высоко их содержание в коже, роговице и конъюнктиве глаз, причем прирост происходит в основном за счет дерматансульфата.
Нами наблюдалось пять больных с синдромом Гурлер. Основные сведения о них представлены в таблице. В качестве примера приводим одно наблюдение.
Катя С., 8 лет, диагноз: мукополисахаридоз I типа (синдром Гурлер). Девочка родилась от второй беременности, протекавшей благоприятно. Мать ребенка до 15-летнего возраста наблюдалась в диспансере по поводу врожденного сифилиса. Психосоматическое развитие ребенка протекало с задержкой, отставание в интеллектуальном развитии стало очевидным после первого года жизни. В последующем ребенок находился в интернате для детей-инвалидов.
При клиническом обследовании обращено внимание на грубые черты лица (нависающий лоб, запавшая переносица, экзофтальм, толстые губы, редкие, дистрофические зубы), кифоз поясничного отдела, деформацию костей предплечья и укорочение пальцевых фаланг, контрактуры суставов, помутнение' роговицы обоих глаз и др.
Со стороны ЦНС отмечались стойкие и значительные изменения, проявившиеся резким снижением интеллекта (IQ = 40 од.) и выраженными общемозговыми расстройствами электрической активности коры головного мозга.
Изменения сердечно-сосудистой системы выражались в резких дистрофических нарушениях миокарда, расширении границ сердца и мезосистолическом шуме. Наряду с этим отмечалось увеличение печени на 5 см, селезенки — па 4 см.
При изучении показателей обмена веществ соединительной ткани установлено увеличение по сравнению с возрастной нормой суточной экскреции с мочой ГАГ в 7 раз (40,1 мг) и снижение в 1,5 раза суточной экскреции ОП (40 мг). Исследование фракционного состава кислых ГАГ мочи позволило идентифицировать преимущественно гепаран- и дерматансульфаты (ГС и ДС).
- Рекомендуем далее ознакомиться со статьей "Мукополисахаридоз II типа (синдром Гунтера) у детей - причины, клиника"
Оглавление темы "Заболевания соединительной ткани у детей":- Болезни соединительной ткани у детей
- Мукополисахаридозы (МПС) у детей - причины, типы
- Мукополисахаридоз I типа (синдром Гурлер) у детей - причины, клиника
- Мукополисахаридоз II типа (синдром Гунтера) у детей - причины, клиника
- Мукополисахаридоз III типа (синдром Санфилиппо) у детей - причины, клиника
- Мукополисахаридоз IV типа (синдром Моркио) у детей - причины, клиника
- Мукополисахаридоз V типа (синдром Шейе) у детей - причины, клиника
- Мукополисахаридоз VI типа (синдром Марото—Лами) у детей - причины, клиника
- Болезнь Марфана у детей - причины, клиника
- Дифференциальная диагностика болезни Марфана