
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Почечно-канальцевый ацидоз (ПКА) - фенотип, диагностика
Почечно-канальцевый ацидоз (ПКА) был впервые описан в 1936 г. и связан с именами Lightwood и Butler. Визуально в клинической картине на первый план выступают костные деформации разной степени тяжести.
На первом году жизни у детей с почечно-канальцевым ацидозом, в отличие от витамин D-дефицитного и витамин D-резистентного рахита, обнаруживаются такие общие симптомы как снижение аппетита, полиурия, полидипсия и быстрая утомляемость.
Задержка физического развития выявляется рано. Рахитоподобные изменения скелета появляются на втором году жизни в виде деформации черепа (лобные и теменные бугры), рахитических «четок» и «браслеток», вальгусной деформации нижних конечностей и мышечной гипотонии. По-существу, эти скелетные изменения ничем не отличаются от других рахитоподобных заболеваний.
Они свидетельствуют о системном остеопорозе, деформациях длинных трубчатых костей, нарушениях костной структуры и др. При морфологических исследованиях костных биоптатов гребешка подвздошной кости обнаруживается уменьшенное количество остеоцитов, изменение костных балок, разрастание нежноволокнистой соединительной ткани.
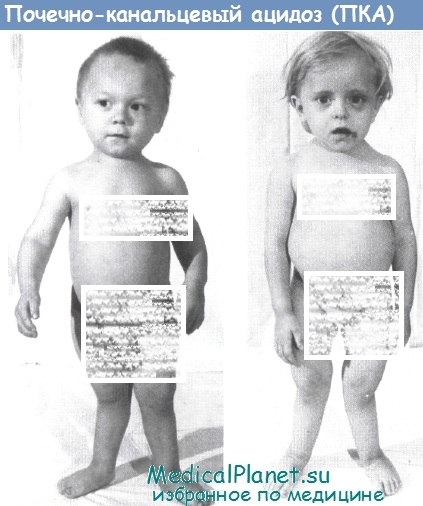
Из литературы известно, что почечно-канальцевый ацидоз по своему происхождению может быть как первичным, таки вторичным. Первичный почечно-канальцевый ацидоз относят к наследственным заболеваниям, а вторичный — обычно связан с сопутствующими болезнями (пиелонефрит, гликогенозы, гипертиреоз и др.).
Выделяют 2 формы почечно-канальцевого ацидоза (ПКА): первичный проксимальный почечный ацидоз, обусловленный врожденным дефектом тубулярной реабсорбции аниона гидрокарбоната, и первичный дистальный почечный тубулярный ацидоз, возникающий в результате неспособности канальцев почек устанавливать адекватный градиент водородных ионов между кровью и тубулярной жидкостью, что и приводит к накоплению водородных ионов в крови и развитию метаболического ацидоза.
Возникающие при этом нарушения фосфорно-кальциевого обмена проявляются в виде гиперкальциурии, снижения почечной экскреции титруемых кислот, аммиака и развития нефрокальциноза. Наряду с этим может отмечаться умеренная протеинурия и лейкоцитурия. На экскреторной урограмме обнаруживаются явления нефролитиаза и нефрокальциноза.
Общие биохимические нарушения при почечно-канальцевом ацидозе (ПКА):
• метаболический ацидоз (дефицит ВЕ= 10-20 ммоль/л);
• умеренная гипофосфатемия (0,9-1,0 ммоль/л);
• гипокальциемия (менее 2,2 ммоль/л);
• повышение активности щелочной фосфотазы;
• низкие показатели мочевой экскреции титруемых кислот (до 25-30 ммоль/сут/ст. пов. тела) и аммиака;
• щелочная или нейтральная реакция мочи (6,7-7,1);
• низкая удельная плотность мочи (1001-1008).
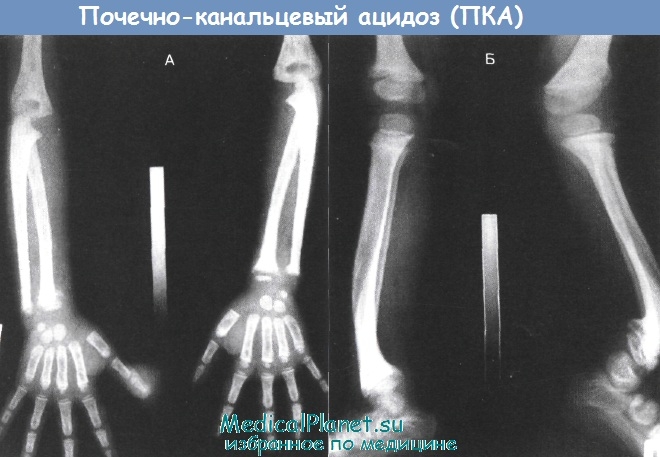
Нарушения структуры костных тканей, системный остеопороз, разрыхление зон препараторного роста, истончение коркового слоя кости.
Клинико-биохимический полиморфизм наследственного ПКА обусловлен генетической гетерогенностью этого заболевания. Описаны случаи аутосомно-рецессивного, аутосомно-доминантного и рецессивного, сцепленного с Х-хромосомой типов наследования. Согласно исследованиям последних лет дистальный почечно-тубулярный ацидоз возникает вследствие G701D мутационных замещений в 1-ом гене, который регулирует хлоридно-бикарбонатные изменения в эритроцитах и клетках дистального нефрона.
В настоящее время выделяют пять клинико-биохимических вариантов наследственного почечного канальцевого ацидоза:
• Почечно-канальцевый ацидоз 1-го типа («Классический» тип. Синдром Лайтвуда-Батлера-Олбрайта), для которого характерен аутосомно-доминантный тип наследования. В основе лежит неспособность дистальных канальцев почек поддерживать градиент водородных ионов между кровью и канальцевым ультрафильтратом. При этом реабсорбция аниона гидрокарбонатов (НСОЗ) в проксимальных канальцах почек не нарушена.
Манифестация заболевания происходит на первом году жизни и носит малоспецифичный характер: периодически возникающая тошнота, рвоты, гипотрофия, гипертермия, задержка статико-моторного развития, запоры, позднее прорезывание зубов и др.
В то же время жажда и полиурия, обнаруживаемые особенно часто, этот симптомокомплекс делают более специфичным и при наличии скелетных деформаций позволяют заподозрить почечно-канальцевый ацидоз (ПКА). Обнаружение нефрокальцино-за при урографическом или же ультразвуковом обследовании в значительной мере укрепляют предположение о наличии почечно-канальцевого ацидоза (ПКА). Окончательным аргументом в пользу этого варианта заболевания служат биохимические изменения.
• Почечно-канальцевый ацидоз 2-го типа (проксимального типа) обычно наследуется по рецессивному, сцепленному с Х-хромосомой, типу. В основе этого варианта болезни лежит нарушение реабсорбции бикарбонатов в проксимальных канальцах почек и их повышенная экскреция с мочой. Манифестация заболевания наступает позже - в возрасте 1,5-3-х лет и тяжесть клинических нарушений менее выражена, чем при ПКА-1. Главным образом она касается снижения темпов роста и психомоторного развития, дефектов зубной эмали.
Биохимические изменения проявляются в виде повышения уровня хлора до 130-135 ммоль/л (при норме 95-107 ммоль/л) и компенсированного метаболического ацидоза. Реакция мочи чаще носит щелочной характер и объясняется повышенной экскрецией анионов гидрокарбонатов.
• Почечно-канальцевый ацидоз 3-го типа (смешанный тип почечного канальцевого ацидоза). Этот вариант встречается редко, тип наследования не установлен, предположительно речь идет об аутосомно-рецессивном типе. В основе заболевания лежит частичное нарушение реабсорбции аниона гидрокарбоната в проксимальных канальцах почек и снижение секреции водородных ионов дистальными канальцами.
Клиническая картина складывается из обычного сочетания рахитоподобных изменений скелета, остеопороза, нефролитиаза, нефрокальциноза, гипокалиемии и мышечной гипотонии. Метаболические расстройства включают: значительный дефицит оснований (ВЕ=-6,0-10,0 ммоль/л), снижение содержания калия, гидрокарбонатов, снижение экскреции титруемых кислот, аммиака, увеличение экскреции гидрокарбонатов, щелочную реакцию мочи.
• Почечно-канальцевый ацидоз 4-го типа (почечно-канальцевый ацидоз с гиперкалиемией). Возникновение этого варианта болезни связано с резистентностью почечных канальцев к действию альдостерона или нарушением его биосинтеза, что сопровождается потерями ионов натрия с мочой, гипонатриемией, гиперкалиемиеи и метаболическим ацидозом.
Заболевание наследуется по аутосомно-рецессивному типу. Для заболевания свойственно: значительная задержка роста, повышенное содержание альдостерона, ренина и калия в плазме при нормальном уровне кортизола в сыворотке крови и 17-ОН и 17 КС в моче. Крайне редко при этом варианте болезни обнаруживается нефрокальциноз.
Учитывая специфику этого варианта дифференциальный диагноз проводится с болезнью Аддисона, дефицитом 21-гидроксилазы, гипоальдостеронизмом, хронической почечной недостаточностью, псевдоальдостеронизмом.
• Почечный канальцевый ацидоз 5-го типа (почечно-канальцевый ацидоз с глухотой). В клинической картине на первый план выступает задержка роста, темпов психомоторного развития и глухота. Кардинальным симптомом при этом является метаболический ацидоз при нормальном уровне калия, щелочной реакции мочи и отсутствии нефрокальциноза.
Основные принципы терапии см. в отдельной статье на нашем сайте МедикалПланет.

П - повышено, С - снижено, N - норма.
- Рекомендуем далее ознакомиться со статьей "Болезнь де Тони-Дебре-Фанкони (ТДФ) - фенотип, диагностика"
Оглавление темы "Наследственные болезни у детей":- Метаболические энцефалопатии детей-подростков: болезнь Шпильмейера-Фогта, детская форма болезни Гоше, семейная полимиоклония
- Метаболические энцефалопатии детей-подростков: болезни Рефсума, Танжера, Андерсона Фабри
- Дифференциация метаболических энцефалопатий детей-подростков
- Принципы диагностики наследственных заболеваний по фенотипу
- Cиндром Лоуренса-Муна-Барде-Бидля (ЛМББ) - фенотип, диагностика
- Фенотипы рахитоподобных заболеваний у детей
- Витамин Д-резистентный рахит (ВДРР) - фенотип, диагностика
- Витамин Д-зависимый рахит (ВДЗР) - фенотип, диагностика
- Почечно-канальцевый ацидоз (ПКА) - фенотип, диагностика
- Болезнь де Тони-Дебре-Фанкони (ТДФ) - фенотип, диагностика