
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Наследственная ксантуренурия (синдром Кнаппа — Комровера) как причина умственной отсталости
В последние годы внимание многих исследователей было обращено на большую группу больных детей с так называемой недифференцированной умственной отсталостью. Высказывалось предположение, что значительная часть этой патологии обусловлена рядом редких рецессивных мутаций и еще не установленных дефектов обмена веществ.
Только при детальном изучении с использованием современных методов биохимического анализа возможно выделение из общей недифференцированной группы умственно отсталых детей с генетически детерминированной патологией. Это предположение оказалось справедливым. Ежегодно в литературе приводятся описания новых, ранее неизвестных форм наследственной патологии (Harris, Hirnschhorn).
В течение нескольких лет нами проводилось изучение состояния здоровья детей, проживающих в условиях северных изолятов (Ю. И. Бараптнев и соавт., Ю. И. Барашпев, Л. З. Казанцева). Это было связано с тем, что для изолированных популяций (отдаленные, труднодоступные маленькие северные села) присуще значительное число родственных браков (инбридинг), в результате чего повышается частота рецессивных мутаций и выщепление гомозиготов по мутантным генам (Н. П. Дубинин, В. П. Эфроимсон, Н. П. Бочков).
При тщательном изучении родословных жителей одного из обследованных сел нам удалось реконструировать родословное древо. Оно представляло собой замкнутый круг, в котором теми или иными родственными узами были связаны 9—10 поколений (более 2500 человек). При этом обнаруживался высокий процент родственных браков (30,5% при норме 1%) и коэффициент инбридинга (F = 0,0042 при норме 0—0,0009).
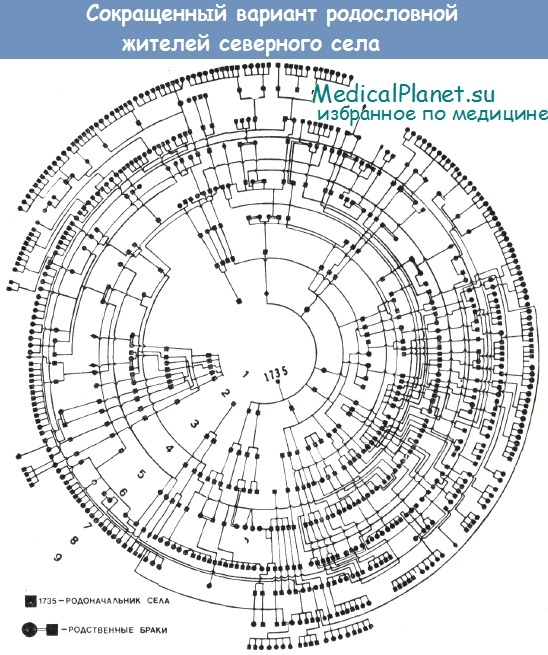
Комплексная оценка состояния здоровья детей показала, что их уровень физического развития и показатели заболеваемости существенно не отличались от возрастных нормативов. Однако среди обследованной нами популяции были выявлены 9 семей, где имелись дети с олигофренией. Случаи заболевания носили семейный характер, наследование осуществлялось по аутосомно-рецессивному типу с полной пенетрантностью патологического признака. При этом олигофрения встречалась в 5 раз чаще в ин-бредных, чем аутбредных, семьях.
При изучении ряда показателей обмена веществ была найдена генерализованная и частично селективная гипераминоацидемия. Заслуживало внимания и то, что аналогичные изменения в обмене обнаруживались не только у больных детей, но и у их фенотипически здоровых родственников, т. е. речь могла идти о наследственных нарушениях обмена веществ.
Полученные нами результаты послужили поводом для последующих исследований. Так, при обследовании детей другого северного села с аналогичным характером родословной, наличием семейных форм олигофрении и генерализованной гипераминоацидемии удалось уточнить характер метаболических нарушений и в первую очередь изменений обмена триптофана и его метаболитов (исследование проведено совместно с Кпарр и Grimm).
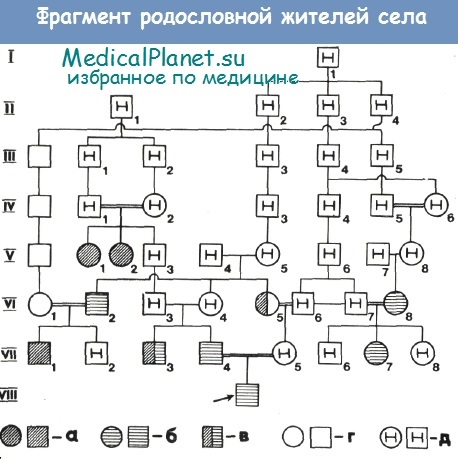
в — умственно отсталые лица с изменениями обмена триптофана; г — здоровые лица без изменений обмена триптофана; д — необследованные лица.
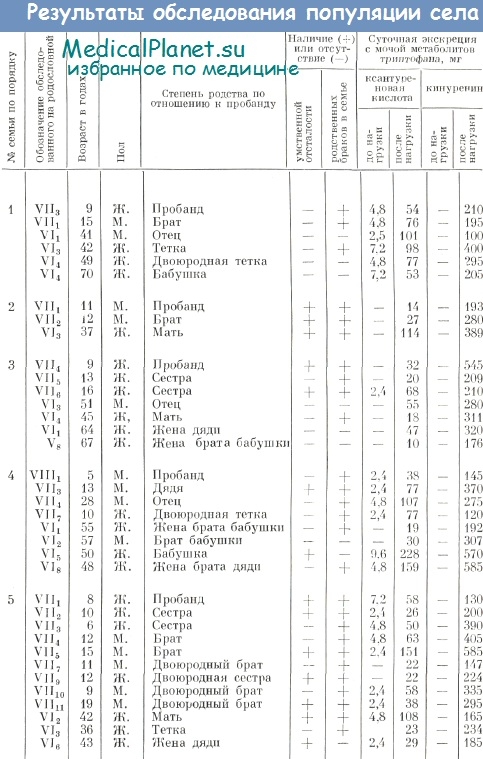
Всего было обследовано 37 человек из 5 семей: 9 умственно отсталых детей, 5 умственно отсталых родственников и 23 феиотипически здоровых родственника. Все больные дети родились от родственных браков. Клиническая картина заболевания была, как правило, однотипна и характеризовалась легкой степенью снижения интеллекта — дебилыюстью. Клинические проявления умственной отсталости становились очевидными с момента начала обучения в школе, когда требовалась мобилизация сложных форм познавательной деятельности, причем заболевание проявлялось в большей степени по мере усложнения школьной программы. Результаты обследования жителей северного села представлены в таблице.
Как видно из таблицы, у подавляющей части обследованных лиц (33 из 37) после нагрузки триптофаном обнаружены высокие величины выведения с мочой кинуренина и особенно ксантуреновой кислоты. Так, очень высокая суточная экскреция с мочой ксантуреновой кислоты (свыше 100 мг) наблюдалась у одного ребенка и 6 взрослых, высокая (от 40 до 100 мг) — у 9 детей и 6 взрослых; высокая суточная экскреция кинуренина (свыше 200 мг) отмечалась у 12 детей и 13 взрослых родственников.
Обращает на себя внимание, что высокие показатели выведения метаболитов триптофана наблюдались как у умственно отсталых, так и у клинически здоровых лиц, однако среди олигофренов отмечались более высокие показатели. Так, среди 9 больных индивидуумов лишь у одного наблюдались нормальные величины экскреции метаболитов триптофана после нагрузки этой аминокислотой, у 4— суточная экскреция с мочой ксантуреповои кислоты превышала 100 мг, а у 4 — колебалась от 40 до 100 мг.
Следует особо отметить высокий удельный вес потомков инбредных браков в изученной популяции (27 из 37), в числе которых были все 18 обследованных детей.
Среди причин, объясняющих обнаруженные метаболические расстройства, прежде всего можно предположить экзогенный дефицит витамина В6, учитывая тот факт, что обследованные лица являлись жителями северного села. В то же время это объяснение представляется мало вероятным, так как витамин В6 содержится во многих пищевых продуктах, особенно в таких, как хлеб, крупы и др., которые потребляются населением в достаточном количестве. Кроме того, обследование проводилось летом, т. е. в период, наиболее благоприятный по потреблению продуктов, содержащих витамин В6.
Аналогичные изменения обмена могли наблюдаться также при употреблении с пищей избыточного количества белка, что приводит к повышенному поступлению триптофана. В связи с этим в организме в больших количествах накапливаются и выделяются с мочой такие его дериваты, как кинуренин, 3-оксикинуренин и 3-оксиантраниловая кислота. Однако при этом не происходит гиперпродукции ксантуреновой кислоты, как это наблюдалось улиц изученной популяции. Кроме того, было установлено, что обследованные нами лица, в особенности дети, получали в сутки не более 150—200 г белка.
Сходные изменения в обмене триптофана могли быть обусловлены также нарушением деятельности эндокринных желез. Известно, что эстрогены угнетают активность кипурениназы и кинуренинтрансаминазы (Rose и соавт.). В то же время глюкокортикоиды и эстрогены активируют триптофанпирролазу. Таким образом, повышенная почечная экскреция исследуемых метаболитов триптофана могла наблюдаться при гиперфункции коры надпочечников или нарушении менструального цикла у женщин. Против первого предположения говорит тот факт, что у обследованных лиц отсутствовали клинические признаки повышенной функции коры надпочечников.
Против второго свидетельствовало то, что среди обследованного контингента были лица обоего пола и дети разного возраста.
Обнаруженные изменения в большей степени соответствовали описанной Knapp в 1958 году витамин В6-зависимой ксантуренурии, при этом лица, имеющие показатели выведения ксантуреновой кислоты после нагрузки свыше 100 мг/сут, могли расцениваться как гомозиготные, а от 40 до 100 мг/сут — как гетерозиготные носители мутаптного гена. Клинические проявления у больных в виде олигофрении степени дебильности могли явиться следствием токсического воздействия на организм высоких концентраций метаболитов триптофана, наряду с возможным недостаточным эндогенным синтезом никотиновой кислоты и ее дериватов, являющихся источником ряда коэнзимов (НАД, НАДФ и др.), обеспечивающих процессы энергетического обмена и необходимых для развивающейся нервной ткани (Tada и соавт., Grimm и соавт.).
Полиморфизм наблюдаемых клинических проявлений мог быть обусловлен различной степенью ферментативной недостаточности (Кпарр, Scriver).
При анализе причин, обусловивших появление витамин В6-зависимой ксантурепурии в изученной популяции, обращает на себя внимание, что большинство обследованных лиц (27 из 37) являлись потомками инбредных браков, причем 28 из 37 сами страдали олигофренией или были ближайшими родственниками умственно отсталых лиц. Семейный характер олигофрении, однотипность клинических и биохимических проявлений заболевания у родственников, высокий удельный вес инбридинга в семьях могут свидетельствовать о том, что выявленная патология имеет генетическое происхождение и связана, по всей вероятности, с длительными процессами инбридинга в селе и «эффектом родоначальника».
- Рекомендуем далее ознакомиться со статьей "Методы изучения обмена триптофана и его метаболитов"
Оглавление темы "Наследственные болезни обмена":- Наследственная ксантуренурия (синдром Кнаппа — Комровера) - клиника, формы
- Диагностика и лечение наследственной ксантуренурии (синдрома Кнаппа — Комровера)
- Пример наследственной ксантуренурии (синдрома Кнаппа — Комровера)
- Нарушение обмена триптофана как причина аллергии у детей
- Наследственная ксантуренурия (синдром Кнаппа — Комровера) как причина умственной отсталости
- Методы изучения обмена триптофана и его метаболитов
- Фенилкетонурия (ФКУ) - клиника, диагностика, пример
- Фенилкетонурия с лейкодистрофией - клиника, диагностика, пример
- Изменения печени при фенилкетонурии - особенности поражения
- Методы диагностики фенилкетонурии