
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Наследственная ксантуренурия (синдром Кнаппа — Комровера) - клиника, формы
Наследственная ксантуренурия (синдром Кнаппа — Комровера) впервые описана Knapp. Впоследствии автор пришел к заключению, что это страдание встречается значительно чаще, чем фенилкетонурия, и обнаруживается у 0,5—1% населения (Knapp).
Клинические проявления наследственной ксантуренурии полиморфны. Вместе с тем при этом заболевании обнаруживается определенный симптомокомплекс — сочетание психоневрологических расстройств с проявлениями аллергии. Изменения центральной нервной системы характеризуются нарушением интеллекта и психики (склонность к истерическим реакциям, состояниям аффекта и немотивированным поступкам, фобии, сомнамбулизм, приступы психомоторного возбуждения, судороги и др.). Полагают, что для наследственной ксантуренурии наиболее типичны изменения интеллекта, так как у большей части наблюдавшихся больных имелась выраженная умственная отсталость (Knapp, O'Brien, Jensen).
Аллергические проявления у больных с синдромом Кнаппа — Комровера характеризуются кожными изменениями (себорейный дерматит, различные экзантемы, фотодерматоз, экзема, нейродермит), бронхиальной астмой и др. Кроме того, как правило, обнаруживается упорный стоматит, хейлит и глоссит.
Родственники пробандов часто страдают такими заболеваниями, как дерматит, бронхиальная астма, новообразования, анемия, расстройства психики, диабет, туберкулез и др. Как сообщают Knapp и Wolfram, обследовавшие родственников больных из 24 семей, у многих из них обнаруживаются обменные нарушения, аналогичные том, которые имелись у пробандов. Обращается внимание на тот факт, что в этих семьях ксантуренурия выявляется также у клинически здоровых лиц.
Таким образом, клиническая симптоматика при наследственной ксантуренурии широко варьирует, и характерные для этого заболевания метаболические расстройства могут обнаруживаться как у больных с выраженным симптомокомплексом, так и у клинически здоровых людей.
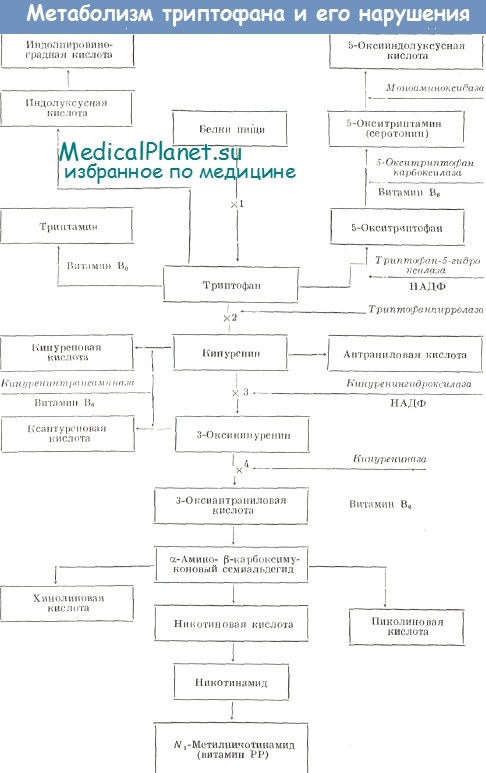
Knapp высказал предположение о том, что клинические проявления расстройства обмена у больных наследственной ксантуренурией связаны, вероятно, с недостаточной активностью пиридоксальзависимой кинурениназы. Результатом этого является избыточное образование таких метаболитов триптофана, как кинуренин, кинуреновая и ксантуреновая кислоты, и недостаточный синтез никотиновой кислоты и ее дериватов.
Исследования, выполненные Tada и соавт. на ткани печени, полученной путем биопсии у детей с пиридоксинзависимой ксантуренурией, показали, что нарушения действительно связаны с дефектом кинурениназы. Активность этого фермента в гомогенате печени детей с ксантуренурией была снижена в 3— 6 раз по сравнению со здоровыми детьми, но нормализовалась при добавлении избыточных количеств экзогенного ПАЛФ. В то же время биосинтез пиридоксаль-5-фосфата у больных не был нарушен, так как содержание его в плазме крови и экскреция с мочой 4-пиридоксиновой кислоты соответствовали норме.
Эти данные свидетельствуют о том, что снижение активности кинурениназы при пиридоксинзависимой ксантуренурии, по-видимому, обусловлено изменением структуры ее белковой части, затрудняющим взаимодействие с коферментом.
Другое объяснение активирующего действия пиридоксаль-5-фосфата может состоять в том, что мутация снижает стабильность апофермента кинурениназы, который при низких концентрациях ПАЛФ быстро теряет свою активность. Повышение концентрации ПАЛФ защищает измененный фермент от инактивации.
Активность других витамин В6-зависимых ферментов (цистатионинсинтетазы и цистатионазы) у больных наследственной ксантуренурией не нарушена.
Связь клинических проявлений с биохимическим дефектом при ксантуренурии до конца не ясна. Полагают, что проявления заболевания обусловлены избыточным образованием ряда метаболитов триптофана (ксантуреновой кислоты, кинуренина и др.) и недостаточным синтезом никотиновой кислоты.
Вместе с тем пиридоксинзависимая ксантуренурия иногда выявляется у вполне здоровых людей и не сопровождается какимилибо клиническими симптомами. Отсутствие типичной клинической картины дает основание некоторым авторам считать пиридоксипзависимую ксантуренуршо безопасным нарушением обмена веществ, а ее сочетание с различными клиническими симптомами — случайным совпадением, в значительной мере обусловленным тем, что исследование обмена триптофана проводилось у лиц с различными заболеваниями. По мнению Knapp, появление различных осложнений при ксантуренурии обусловлено действием дополнительных экзогенных и генетических факторов. Возникающий в связи с этим вопрос о целесообразности обязательной коррекции ксантуренурии путем регулярного приема повышенных доз пиридоксина, в том числе и клинически здоровыми людьми, требует тщательного изучения и длительных наблюдений.
Полиморфизм клинических проявлений заболевания, как полагают, обусловлен различной степенью ферментативной недостаточности. Так, активность печеночной кинурениназы у двух больных детей была различной, что находило отражение не только в различных количественных показателях, но и в неодинаковой чувствительности к активирующему действию экзогенного пиридоксальфосфата (Tada и соавт.).
Выраженность биохимических изменений, затрагивающих обмен триптофана у больных наследственной ксантуренурией, также может быть различной, так как в ряде случаев активность кинурениназы практически отсутствует.
Примером такой формы может, по-видимому, служить оксикинуренинурия, описанная Komrower и соавт., — врожденное нарушение обмена триптофана на пути его превращения в никотиновую кислоту, при котором, как и в случае пиридоксинзависимой ксантуренурии Кнаппа, экскретируются повышенные количества кинуренина, 3-оксикинуренина и ксантуреновой кислоты. Однако это повышение наблюдается на обычной диете, без нагрузки триптофаном, и не корригируется даже при регулярном приеме пиридоксина.
В описанном Komrower и соавт. случае девочка 14 лет наблюдалась с трехнедельного возраста по поводу стоматита, гемолитической анемии и энтероколита. С шестинедельного возраста ребенок регулярно получал поливитамины, в том числе 5 мг никотипамида, что могло быть одним из факторов, способствовавших клиническому улучшению. При прекращении приема витаминов развивалась повышенная чувствительность кожи к солнечному свету (фотодерматоз), которая проходила при назначении иикотинамида. Хроматографическое изучение обмена триптофана в 9-летном возрасте выявило повышенную экскрецию 3-оксикипурепина и ксаитуреповой кислоты, превышавшую порму в 30—60 раз и еще более резко возраставшую после нагрузки триптофаном. Экскреция кинуренина и кипуреновой кислоты при нагрузке триптофаном превышала норму в 10—20 раз. В то же время в моче отсутствовали антрапиловая и 3-оксиантраниловая кислоты. Эти парушения свидетельствовали о наличии блока в обмене триптофана на уровпе кинурениназы. Существование этого блока почти полностью нарушало эндогенное образование никотиновой кислоты из триптофапа. Об этом свидетельствовало отсутствие увеличения экскреции N-метилпиридона и N-метшпшкотипамида после нагрузки триптофаном. По мнению авторов, описавших этот случай, клинические проявления заболевания могли быть обусловлены токсическим действием накапливающихся продуктов обмена триптофана и нарушелием эндогенного синтеза никотиновой кислоты.
По локализации биохимического дефекта — на уровне кинурениназы — оксикинуренинурия, описанная Komrower и соавт., идентична ксантуренурии Кнаппа, однако в отличие от последней она не корригируется повышенными дозами пиридоксина, что указывает на иной молекулярный механизм нарушения. Если при пиридоксипзависимой ксантуренурии активность кипурениназы снижается в 3—6 раз по сравнению с нормой и механизм нарушения, по-видимому, состоит в изменении структуры апофермента, затрудняющем взаимодействие с ПАЛФ, то при оксикинуренинурии Комровера активность кипурениназы, вероятно, отсутствует практически полностью.
Таким образом, ксантуренурию Кнаппа и оксикинуренинурию Комровера, по-видимому, можно рассматривать как пиридоксинзависимую и пиридоксинрезистентную формы врожденной ксантуренурии. Полагают, что витамин В6-зависимая ксантуренурии наследуется по аутосомно-рецессивному типу.
- Рекомендуем далее ознакомиться со статьей "Диагностика и лечение наследственной ксантуренурии (синдрома Кнаппа — Комровера)"
Оглавление темы "Наследственные болезни обмена":- Наследственная ксантуренурия (синдром Кнаппа — Комровера) - клиника, формы
- Диагностика и лечение наследственной ксантуренурии (синдрома Кнаппа — Комровера)
- Пример наследственной ксантуренурии (синдрома Кнаппа — Комровера)
- Нарушение обмена триптофана как причина аллергии у детей
- Наследственная ксантуренурия (синдром Кнаппа — Комровера) как причина умственной отсталости
- Методы изучения обмена триптофана и его метаболитов
- Фенилкетонурия (ФКУ) - клиника, диагностика, пример
- Фенилкетонурия с лейкодистрофией - клиника, диагностика, пример
- Изменения печени при фенилкетонурии - особенности поражения
- Методы диагностики фенилкетонурии