
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии новорожденных: алкаптонурия, аргининемия, аргинин-янтарная ацидурия, гиперлизинемия
В группу наследственных метаболических энцефалопатий периода новорожденности входят наследственные заболевания обмена аминокислот. Аминокислотам принадлежит огромная роль в развитии и становлении организма ребенка. Они являются основными структурными элементами белков, используются для синтеза гормонов, иммунных тел, служат источником энергии и др. При нарушениях метаболизма аминокислот возникает реальная опасность для здоровья и жизни ребенка.
Наследственные болезни аминокислот составляют наиболее обширную группу заболеваний, они относятся к генетически детерминированным ферментопатиям, носят моногенный характер и имеют аутосомно-рецессивный тип наследования. К самым частым заболеваниям этой группы относится ФКУ (1:10 000).
Вся сложность своевременной диагностики наследственных заболеваний обмена аминокислот состоит в том, что большая часть из них не имеет специфических черт. Возникающие поражения нервной системы, кожи и других органов часто считают последствиями внутричерепных родовых травм, менингитов и энцефалитов и т.п. Окончательный диагноз становится возможным только после проведения специальных исследований обмена аминокислот и определения ферментов.
В практической деятельности далеко не всегда удается осуществить диагностику всех описанных в литературе нозологических форм этой группы заболеваний. Диагностический процесс по своей сложности иногда напоминает научно-исследовательскую работу. Мы считаем необходимым ограничить перечень заболеваний аминокислотного обмена, предоставив критерии дифференциальной диагностики только для 26 из них.
1. Алкаптонурия является первым заболеванием, описанным Гарродом, как врожденное нарушение метаболизма из-за дефицита фермента гомогентизиноксидазы. Мутированный ген картируется на хромосоме 3. Биохимически происходит накопление гомогентизиновой кислоты в тканях и в биологических жидкостях.
У новорожденных намокшие пеленки при взаимодействии с воздухом быстро приобретают темный красно-коричневый цвет. В последующем окрашиваются склеры и слизистые оболочки, развиваются охронозные артриты. Влечении главным является назначение диеты с низким содержанием фенилаланина и тирозина.
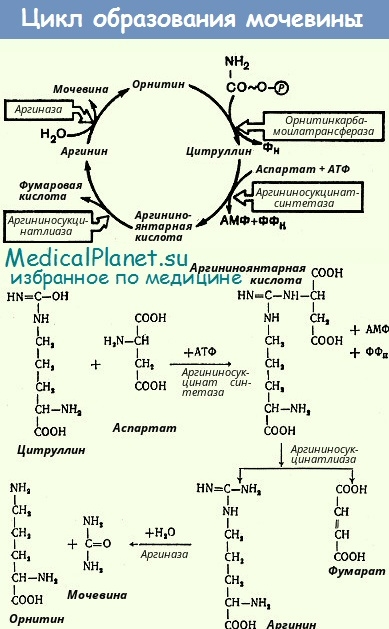
2. Аргининемия - задержка психомоторного развития, эпизоды гипераммониемии, рвота, мышечная гипертония, парезы и параличи, судороги, гепатомегалия, угнетение ЦНС, атаксия, дефицит массы тела. Дефект фермента аргиназы. Биохимические изменения: высокие уровни аргинина в крови и моче, аммиака в крови.
3. Аргинин-янтарная ацидурия - задержка психомоторного развития, судороги, измененные волосы (ломкость и сухость), мышечная гипотония, атаксия, гепатомегалия, дефицит массы тела, респираторные расстройства, рвота. Дефект фермента аргининсукциназы. Биохимические изменения: высокий уровень аргинин-янтарной кислоты в крови и моче и аммиака в крови.
4. бета-Метилкротонилглицинурия - измененный запах мочи (запах кошачьей мочи), упорная рвота, кетоацидоз, судороги, мышечная гипотония, респираторные расстройства, дефицит массы тела. Дефект фермента бета-метилкротонил КоА-карбоксилазы. Гиперлейцинемия, в моче повышена экскреция бета-метилкротонилглицина бета бета-гидрооксиизовалериановой кислоты.
5. Гиперлизинемия - врожденный дефект обмена, связанный с дефицитом ферментов лизин-кетоглутаратовой редуктазы и сахоропиндегидрогеназы. Гены картированы в локусе LYS1 и LYS 9. Наследование аутосомно-рецессивное. Выделяют два типа гиперлизинемии:
Гиперлизинемия I типа: упорная рвота, судороги, задержка психомоторного развития, угнетение ЦНС, мышечная гипотония.
Гиперлизинемия II типа- те же симптомы + низкий рост, дефицит массы тела, слабость связочного аппарата, гепатомегалия, тугоухость, истонченная кожа, костные деформации. Биохимические изменения: гиперлизинемия, гиперлизинурия.
6. Гиперпипеколатемия - гепатомегалия, задержка психомоторного развития, рвота, мышечная гипотония парезы и параличи, изменения на глазном дне. Дефект фермента неизвестен. В моче и крови повышен уровень пипеколовой кислоты, генерализованная гипераминоацидурия.
- Рекомендуем далее ознакомиться со статьей "Гистидинемия - клиника, диагностика"
Оглавление темы "Наследственная патология":- Синдром Эллерса-Данлоса - клиника, диагностика
- Наследственные метаболические энцефалопатии - причины, классификация
- Метаболические энцефалопатии новорожденных: алкаптонурия, аргининемия, аргинин-янтарная ацидурия, гиперлизинемия
- Гистидинемия - клиника, диагностика
- Метаболические энцефалопатии новорожденных: гомоцистинурия, D-глицериновая ацидемия, изовалериановая ацидемия, карнозинемия
- Лейциноз - клиника, диагностика
- Метаболические энцефалопатии новорожденных вследствие недостаточности ферментов
- Тирозинемия - клиника, диагностика
- Обследование при метаболических энцефалопатиях новорожденных
- Симптомы нарушения обмена аминокислот у новорожденных в таблице