
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии детей ясельного возраста: метахроматическая лейкодистрофия
К наследственным метаболическим энцефалопатиям детей ясельного возраста относятся заболевания, манифестация которых происходит несколько позднее. Первые признаки обнаруживаются на 1-4-м годах жизни. К этому времени дети уже могут самостоятельно ходить, говорить, активно относиться к окружающему миру. В связи с этим утрата приобретенных ими навыков становится настолько очевидной, что может служить для врача важным признаком в процессе дифференциальной диагностики.
Все заболевания этой группы, несмотря на их разный генез, объединяет пять общих признаков: прогрессирующая параплегия; неустойчивая походка и некоординированные движения; судорожные состояния и миоклония (иногда атаксия); задержка или регресс психических функций; интермиттирующие неврологические расстройства (ступор; кома, измененное поведение, атаксия, судороги).
Для заболеваний этой группы свойственно также сочетание выраженных изменений ЦНС с висцеральными расстройствами, изменениями скелета, зрения и др.
К наиболее важным симптомам относятся:
• неврологические расстройства (полиневриты, пирамидные и бульбарные нарушения, демиелинизация);
• глазные симптомы (нарушение функции глазодвигательных нервов, поражение сетчатой оболочки глаза, конъюнктивит, снижение зрения вплоть до слепоты);
• глухота;
• измененный размер головы (микро- или макроцефалия).
Неврологические расстройства и интеллектуальный регресс хотя и обнаруживаются рано, тем не менее достигают своего максимума спустя 2-4 года и позже.
По особенностям манифестации могут быть выделены: заболевания, при которых доминируют двигательные расстройства (парапарез, атаксия или атетоз); заболевания, начинающиеся с миоклонуса и судорог; заболевания, сопровождающиеся задержкой психомоторного развития и изменения многих органов и систем.
Метахроматическая лейкодистрофия (болезнь Шольца-Гринфильда, детский сульфатидоз)
Манифестация на 2-м году жизни: прогрессирующие нарушения двигательных функций в различных комбинациях: вялый парапарез с гипотонией, отсутствие сухожильных рефлексов; сочетание пирамидных знаков (спастика, рефлексы Бабинского и Россолимо) и глубокое угнетение сухожильных рефлексов; спастическая параплегия с повышением сухожильных рефлексов.
Метахроматическая лейкодистрофия (болезнь Шольца-Гринфильда, детский сульфатидоз) быстро прогрессирует и в дальнейшем характеризуется спастическим тетрапарезом, атактическим синдромом, возможны судороги и гиперки-незы. В последующем снижается интеллект, наступают расстройства речи, зрения и слуха, появляются бульбарные и псевдобульбарные симптомы, децеребрационная ригидность, макроэнцефалия.
В спинномозговой жидкости обнаруживается белково-клеточная диссоциация, в плазме крови - повышенное содержание сульфатидов. Смерть наступает спустя 2-3 года от начала заболевания. Патоморфологически определяется диффузная симметричная демиелинизация пирамидных, мозжечковых путей и путей проприоцептивной чувствительности. В основе заболевания дефицит арилсульфатазы А. Тип наследования — А/Р. Дифференциальный диагноз проводится с адреномиелоневропатией, семейной спастической параплегией с лейкодистрофией, церебральным параличом или церебральной спастической параплегией Штрюмпеля, врожденной миопатией или миелопатией, болезнью Съёгрена-Ларсена, болезнью Аустина и нейроаксональной дегенерацией.
Метахроматическая лейкодистрофия в сочетании с фенилкетонурией - очень редкий вариант наследственной патологии. Дети к 2-м годам жизни утрачивают ранее приобретенные навыки. Специфические фенотипические признаки (светлые волосы, голубые глаза, белая депигментированная кожа), а также умственная отсталость позволяют подозревать ФКУ. Действительно, концентрация фенилаланина (ФА) в сыворотке крови может достигать 39,5мг%. Активность фенилаланингидроксилазы в печени оказывается резко сниженной (до 0,014 мкМ тирозина на 1 г белка за 1 час при норме 0,8-2,4 мкМ тирозина на 1 г белка за 1 час). При обследовании родителей ребенка они оказываются гетерозиготны по гену ФКУ.
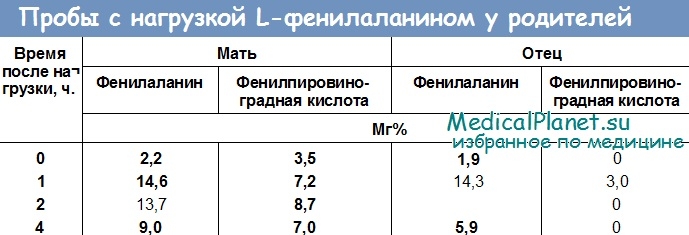
Особенно высокая экспрессивность гена ФКУ может быть обнаружена у матери ребенка, у которой еще до нагрузки L-фенилаланином в моче обнаруживается высокое содержание фенилпировиноградной кислоты (3,5 мг%). После нагрузки L-фенилаланином (0,1 на 1 кг массы тела) у обоих родителей спустя 60 и 120 минут отмечается повышенный уровень ФА крови, не приходящий к норме спустя 4 часа. Более того, у обоих родителей в процессе нагрузки наблюдается значительное увеличение экскреции фенилпировиноградной кислоты.
Несмотря на нормализацию обмена фенилапанина на фоне длительного лечения, состояние ребенка продолжает ухудшаться: потеря зрения, децеребрационная ригидность, тонические судороги, нарушается акт жевания и глотания, атрофия зрительных нервов. Активность арилсульфатазы А в лейкоцитах и плазме крови резко снижается, процент положительно реагирующих клеток при окрашивании толуидиновым синим оказывается равным 39, при норме 25,7, а цитохимический коэффициент 0,41 при норме 0,24.
- Рекомендуем далее ознакомиться со статьей "Метаболические энцефалопатии детей ясельного возраста: болезнь Аустина, нейроаксональная дегенерация, синдром Луи Бара"
Оглавление темы "Метаболические энцефалопатии у детей":- Метаболические энцефалопатии детей ясельного возраста: метахроматическая лейкодистрофия
- Метаболические энцефалопатии детей ясельного возраста: болезнь Аустина, нейроаксональная дегенерация, синдром Луи Бара
- Метаболические энцефалопатии детей ясельного возраста: болезни Нимана-Пика, Дери, Краббе, Гоше
- Метаболические энцефалопатии детей ясельного возраста: нейрональный цероидлипофусциноз, синдром Альперса
- Дифференциация метаболических энцефалопатий детей ясельного возраста
- Метаболические энцефалопатии детей-подростков: болезнь Вильсона-Коновалова, хорея Гентингтона, синдром Ханта
- Метаболические энцефалопатии детей-подростков: прогрессирующая торсионная дистония, болезнь Галлервордена-Шпатца, синдром хореоатетоза
- Метаболические энцефалопатии детей-подростков: атрофия Corpus Luisi, лейкодистрофии
- Метаболические энцефалопатии детей-подростков: атаксия Фридрейха, синдромы Русси-Леви, Ван-Богарта-Шредера-Эпштейна
- Метаболические энцефалопатии детей-подростков: синдром Базена-Корнцвейга, миоклоническая эпилепсия типа Лафора