
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии детей ясельного возраста: болезни Нимана-Пика, Дери, Краббе, Гоше
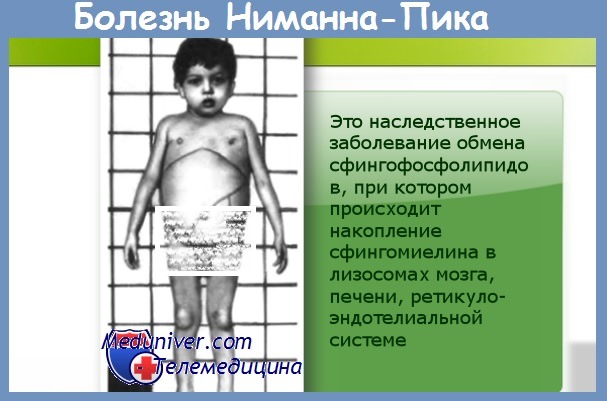
1. Поздняя детская и юношеская хроническая болезнь Нимана-Пика (болезнь Нимана-Пика, типы С и D). Эти варианты болезни отличаются оттипов А и В временем манифестации, течением патологического процесса и тяжестью нейровисцеральных нарушений. Манифестация варианта С происходит на 2-4-м году жизни, а варианта D - на 5-8-м.
Среди неврологических симптомов: прогрессирующая деменция, дизартрия, церебеллярная атаксия, частые судороги, экстрапирамидные знаки, гепатоспленомегалия, офтальмоплегический синдром (атрофия сосков зрительного нерва, «вишневая косточка» на глазном дне). В основе заболевания - нарушения в обмене сфингомиелина за счет инактивации сфингомиелиназы. Тип наследования — А/Р.
2. Поздний детский Gm1-ганглиозидоз, тип II (болезнь Дери). Манифестация болезни происходит на втором году жизни (в 12-18 месяцев). Отмечаются затруднения при ходьбе, неловкая походка, локомоторная атаксия, мышечная гипотония, повышение сухожильных рефлексов. Болезнь быстро прогрессирует: снижается интеллект, нарастает атаксия, спастический тетрапарез, появляются клонические судороги. Смерть наступает в возрасте 8-10 лет. В костном мозге и в тканях внутренних органов обнаруживаются «пенистые клетки».
В белом веществе мозга выражена демиелинизация. Gm1-ганглиозиды накапливаются только в ткани головного мозга. Заболевание отнесено к группе муколипидозов. Оно обусловлено дефицитом изоферментов В и С, бета-галактозидазы. Тип наследования — А/Р. Дифференциальный диагноз проводится со всеми прогрессирующими энцефалопатиями, которые начинаются на втором году жизни и сопровождаются двигательными расстройствами (метахроматическая лейкодистрофия, Gm2-ганглиозидоз, нейроаксональная дегенерация и пр.).
3. Gm1-ганглиозидоз, тип III. Манифестация заболевания во второй половине первого года: генерализованные судороги, мышечная гипертония, гепатоспленомегалия, в последующем спастический тетрапарез. В костном мозге обнаруживаются вакуолизированные лимфоциты, в моче повышено содержание нецитилпиридиновых гликопротеинов, преципитируемых хлоридом. В основе заболевания - снижение активности бета-галактозидазы.
4. Поздняя детская и юношеская лейкодистрофия Краббе (глобоидноклеточная лейкодистрофия с поздним началом). Манифестация заболевания на 2-6-м году жизни: повышение мышечного тонуса в нижних конечностях и в разгибателях спины, атаксия, дизартрия, снижение интеллекта, судороги, атрофия зрительных нервов. В спинномозговой жидкости повышен уровень белка. Смерть наступает через 1-6 лет от начала заболевания.
Гистологически обнаруживаются сегментальная демиелинизация, астроцитарный глиоз, в ткани мозга накапливается церамиддигексозид. Предполагают, что в основе заболевания лежит снижение активности бета-галактозидазы цереброзидов. Тип наследования —А/Р.
5. Поздняя детская и юношеская формы болезни Гоше (болезнь Гоше, тип III). К 6-8 годам уже имеются тяжелые церебральные нарушения: атаксия, спастика, спленомегалия, скелетные аномалии. Зрение остается неизменным. В последующем появляются умственная отсталость, изменения поведения, двигательные расстройства. Патоморфологически отмечается резкое увеличение селезенки. В тканях печени, селезенки, мозга и других органах обнаруживают клетки Гоше. В основе заболевания -дефицит глюкоцереброзидазы.
- Рекомендуем далее ознакомиться со статьей "Метаболические энцефалопатии детей ясельного возраста: нейрональный цероидлипофусциноз, синдром Альперса"
Оглавление темы "Метаболические энцефалопатии у детей":- Метаболические энцефалопатии детей ясельного возраста: метахроматическая лейкодистрофия
- Метаболические энцефалопатии детей ясельного возраста: болезнь Аустина, нейроаксональная дегенерация, синдром Луи Бара
- Метаболические энцефалопатии детей ясельного возраста: болезни Нимана-Пика, Дери, Краббе, Гоше
- Метаболические энцефалопатии детей ясельного возраста: нейрональный цероидлипофусциноз, синдром Альперса
- Дифференциация метаболических энцефалопатий детей ясельного возраста
- Метаболические энцефалопатии детей-подростков: болезнь Вильсона-Коновалова, хорея Гентингтона, синдром Ханта
- Метаболические энцефалопатии детей-подростков: прогрессирующая торсионная дистония, болезнь Галлервордена-Шпатца, синдром хореоатетоза
- Метаболические энцефалопатии детей-подростков: атрофия Corpus Luisi, лейкодистрофии
- Метаболические энцефалопатии детей-подростков: атаксия Фридрейха, синдромы Русси-Леви, Ван-Богарта-Шредера-Эпштейна
- Метаболические энцефалопатии детей-подростков: синдром Базена-Корнцвейга, миоклоническая эпилепсия типа Лафора