
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Клиника и проявления Лоуренса—Муна—Барде—Бидля (ЛМББ)
В процессе изучения синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ) неоднократно предпринимались попытки систематизации имеющихся в литературе данных. Так, в 1937 году Warcani, проанализировав литературные данные о 102 больных с синдромом ЛМББ, пришел к выводу, что наиболее частым признаком заболевания является ожирение (76 больных из 102), затем — пигментный ретинит ((71), поли- и синдактилия (70), умственная отсталость (64), генитальная дистрофия (59 больных). Наличие в клинической картине всех 5 признаков обнаружено только у 24 из 102 больных.
Это привело автора к мысли о том, что далеко не всегда для данного заболевания характерен полный набор симптомов и что отсутствие некоторых из них может приводить к неправильной оценке состояния больных и поздней диагностике синдрома ЛМББ. Это совпадает с мнениями Blummel и Kniker, проанализировавших литературу 1949—1959 годов. Таким образом, подчеркивается клинический полиморфизм синдрома ЛМББ и выраженная вариабельность отдельных его симптомов.
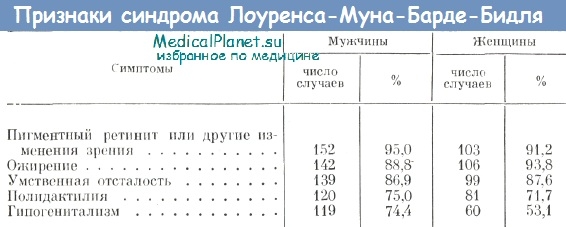
Наибольший интерес может представлять фундаментальное исследование Bell, изучившей родословные 368 больных, ранее описанных в литературе. Обобщение большого литературного материала позволило автору определить информативную ценность кардинальных признаков синдрома ЛМББ. Особенно высока диагностическая ценность таких симптомов, как пигментный ретинит (встречаемость более 90%), затем ожирение (около 90%), умственная отсталость, полидактилия и гипогенитализм. Следует, однако, заметить, что клинические проявления кардинальных симптомов заболевания обычно отличаются определенной пестротой.
Нарушения зрения выражаются не только пигментным ретинитом, но и атипичной мелкоточечной ретинопатией, точечным альбосцентным ретинитом, макулярной дегенерацией, экстраренальной офтальмоплегией, миопией, микрофтальмией, аниридией и пр.
Снижение остроты зрения обычно начинается с сужения полей и очень быстро прогрессирует, приводя нередко к амаврозу. В исследованиях Klein и Amman 73,3% больных были слепыми уже к 20 годам, а 86,4% —в возрасте 30 лет. Обычная пигментная дегенерация сетчатки ведет к слепоте в возрасте от 40 до 50 лет.
По мнению Ehrenfeld и соавт., изменения глазного дна у больных следует рассматривать в эволюционном аспекте, так как нехарактерные изменения при первом обследовании позднее, с возрастом, могут стать типичными. Ehrenfeld и соавт. и Stanescij указывают, что офтальмоскопически пигментный ретинит ярко выражен у 15% больных, в то время как ночной слепотой страдают все больные.
В случаях типичного пигментного ретинита при офтальмоскопии на сетчатке отмечается скопление пигмента в виде «костных телец» по периферии и в макулярной области, восковидная окраска диска зрительного нерва с наличием белоточечного поражения макулярной области, сужение сосудов.
При гистологическом исследовании глаз обнаруживается атрофия палочек и колбочек с пролиферацией пигментного эпителия, проникающего в склеру, изменение ретинальных и хориоидальных сосудов, гибель нейроэпителия и ганглиозных клеток по всей сетчатой оболочке.
Второй кардинальный симптом синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ) - ожирение - также крайне вариабелен в своих проявлениях: от I—II до III—IV степени. Чрезмерная масса тела у больных обнаруживается обычно уже на первом году жизни и в последующем быстро прогрессирует. У детей с синдромом ЛМББ нередко отмечается снижение толерантности к глюкозе и основного обмена. В семьях больных часто встречаются лица, страдающие ожирением или сахарным диабетом.
Результаты собственных исследований и данные литературы свидетельствуют также о большой вариабельности клинической картины умственной отсталости у больных с синдромом ЛМББ: от легкой дебильности до идиотии (В. М. Пахомова). Нами наблюдалось 28 больных с синдромом ЛМББ. Комплексные обследования с использованием клинических и нейрофизиологических методов позволило установить, что ЦНС почти всегда в той или иной степени вовлекается в патологический процесс.
К наиболее частым симптомам поражения нервной системы можно отнести: задержку психомоторного развития в раннем детстве; умственную отсталость (DQ=18—57 ед. при норме выше 80 ед.); нарушения биоэлектрической активности головного мозга (дизритмия, снижение вольтажа и реакции на афферентные раздражители); изменения Эхо-ЭГ (расширение III желудочка мозга, увеличение индекса боковых желудочков мозга, внутричерепная гипертензия).
Однако выраженность психоневрологических расстройств неодинакова при разных вариантах болезни. Так, у детей с абортивной формой синдрома ЛМББ интеллект обычно соответствует возрастной порме, у детей же с полным синдромом ЛМББ речь идет обычно о тяжелой умственной отсталости. Особенности интеллектуального дефекта при клинических вариантах синдрома ЛМББ представлены в таблице.
Большинство исследователей отмечают, что умственная отсталость у больных нередко сочетается с расстройствами речи и слуха, гиперкинезами, явлениями паркинсонизма, эпилепсией и другой неврологической симптоматикой.
Сведения о морфологии головного мозга при синдроме Лоуренса—Муна—Барде—Бидля (ЛМББ) крайне скудны. Известно всего 12 случаев аутопсий. При гистологическом изучении отмечался дегенеративный процесс в ядрах гипоталамуса, глиозные разрастания, кистевидные изменения передней и задней доли гипофиза, иногда расширение III желудочка мозга.
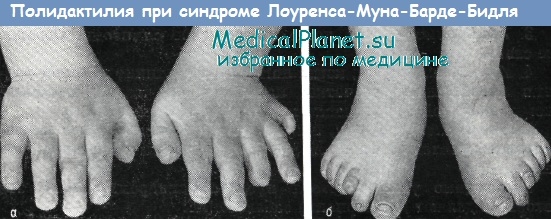
Полидактилия также относится к часто встречаемым при синдроме Лоуренса—Муна—Барде—Бидля (ЛМББ) признакам. Обычно обнаруживается шестипалость, проявляющаяся с латеральной стороны кисти или стопы. Добавочный палец, как правило, гипопластичен, имеет форму отростка. Иногда полидактилия обнаруживается только рентгенологически. Часто как эквивалент полидактилии отмечается синдактилия или брахидактилия.

По данным Klein и Amman, брахидактилия встречается чаще, чем другие изменения конечностей (в 83,0% случаев), что же касается синдактилии, то она часто бывает кожного типа.
Гипогенитализм по сравнению с другими симптомами болезни обнаруживается несколько реже. У лиц мужского пола этот признак выявляется относительно просто: евнухоидный тип телосложения, иногда гинекомастия, рост волос на лобке по женскому типу, одно- или двусторонний крипторхизм, тестикулярная гипоплазия, малые размеры полового члена, недоразвитие мошонки, снижение и полное отсутствие libido sexualis. У женщин выявление полового инфантилизма представляет большие трудности.
Обычно принимаются во внимание: недоразвитие вторичных половых признаков, нарушение менструального цикла (олиго- или аменорея), атрезия влагалища, гипоплазия яичников и др. Тем не менее в литературе имеются описания, когда женщины с синдромом ЛМББ становились матерями, а мужчины вступали в брак и имели детей. При изучении гормонального профиля (17-кетостероидов и 17-оксикетостероидов) не было выявлено каких-либо четких изменений. У больных обнаруживалась нормальная, сниженная или повышенная функция надпочечпиков. Морфологическое исследование яичек у больных также не позволило получить достоверных сведений об их изменении, высказывалось лишь мнение о нарушении сперматогенеза из-за аплазии этих желез. В яичниках у женщин были найдены дистрофические изменения.
Помимо основных пяти симптомов, характерных для синдрома ЛМББ, у больных нередко могут наблюдаться и другие изменения. Прежде всего это врожденные пороки скелета, сердца, желудочно-кишечного тракта, кожи и др. При обследовании детей старшего возраста может быть впервые диагностирован вторичный пиелонефрит, возникший на почве таких аномалий развития, как поликистоз почек, гидронефроз, расширение мочеточников и др.
Таким образом, синдром Лоуренса—Муна—Барде—Бидля (ЛМББ) относится к заболеваниям с яркой клинической картиной, и его своевременная диагностика, казалось, не представляет значительных трудностей. Однако опыт показывает, что диагноз этого заболевания ставится крайне редко, а большинство больных наблюдаются у врачей по поводу самой разнообразной патологии. Так, если в клинической картине превалирует ожирение, больные состоят на учете у эндокринолога, при доминировании нарушений зрения — у офтальмолога, при умственной недостаточности — у психиатра и т. п.
Трудность диагностики синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ) объясняется прежде всего клиническим полиморфизомом и прогредиентным течением заболевания. Klein и Amman, подчеркивая клиническое разнообразие этого синдрома, предлагают пользоваться следующей классификацией:
1. Полная форма синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ) — при наличии 5 кардинальных признаков.
2. Неполная форма, когда отсутствуют один или два ведущих симптома (например, полидактилия или гипогенитализм).
3. Абортивная форма, когда клинические симптомы выражены слабо.
4. Атипичная форма, когда пигментный ретинит заменен другими поражениями сетчатки, например оптической атрофией, экстраренальной офтальмоплегией, высокой миопией и др.
5. Экстенсивная форма, при которой в добавление к 5 кардинальным симптомам имеются другие врожденные аномалии развития.
Необходимо указать, что в пределах синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ) эти авторы выделяют два варианта заболевания:
1. Синдром Бимонда, описанный отдельно датским невропатологом в 1934 году и характеризующийся коломобой радужной оболочкой, гипофизарным инфантилизмом, полидактилией, умственной отсталостью.
2. Синдром Альстрема — Хальгрена — Эсандера, описанный в 1959 году, при котором имеется сочетание ретинопатии, ожирения, диабета, глухоты и полидактилии.
Из 57 больных с синдромом Лоуренса—Муна—Барде—Бидля (ЛМББ), обнаруженных Klein и Amman, 26 имели классическую клиническую картину, 11 — неполную, 6 — абортивную, 5 — атипичную, а 9 больных были с неопределенным типом заболевания. Заслуживает внимания мнение Blurnmel и Kniker, согласно которому присутствие всех признаков синдрома ЛМББ не только не обязательно, но даже маловероятно, так как ген, ответственный за любой симптом, проявляет свое действие во взаимосвязи с другими генами, что может приводить к возникновению клинических вариантов синдрома.
Поздняя диагностика синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ) обусловлена также тем, что при рождении ребенок не имеет какой-либо специфической клинической картины. Единственным симптомом может быть лишь полидактилия. Только к концу первого года жизни становится очевидным склонность к ожирению. Снижение зрения чаще всего наблюдается в возрасте 4 и более лет, а умственная отсталость — в дошкольном возрасте. Таким образом, полный симптомокомплекс может обнаружиться только в возрасте 8—10 лет и старше.

- Рекомендуем далее ознакомиться со статьей "Пример синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ)"
Оглавление темы "Патология обмена липидов у детей":- Критерии диагностики гиперлипопротеидемии I типа у детей
- Пример гиперлипопротеидемии I типа у ребенка
- Синдром Лоуренса—Муна—Барде—Бидля (ЛМББ) - причины, эпидемиология
- Клиника и проявления синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ)
- Пример синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ)
- Диагностика и дифференциация синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ)
- Лечение синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ)
- Методы диагностики синдрома Лоуренса—Муна—Барде—Бидля (ЛМББ)
- Компоненты соединительной ткани и их значение
- Метаболиты соединительной ткани и их оценка