
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Метаболические энцефалопатии детей-подростков: прогрессирующая торсионная дистония, болезнь Галлервордена-Шпатца, синдром хореоатетоза
1. Прогрессирующая торсионная дистония. Манифестация заболевания в возрасте 10-13 лет: вращательные спазмы туловища, шеи, проксимальных отделов конечностей, деформации позвоночника, кривошея, интеллект сохранен. При патоморфологическом исследовании обнаруживают изменения в чечевичных ядрах, красных ядрах и люисовом теле. Патогенез неясен. Тип наследования — А/Д.
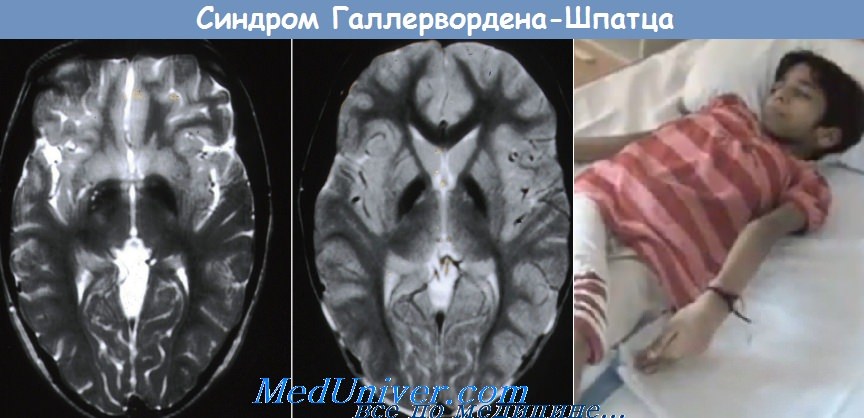
2. Болезнь Галлервордена-Шпатца. Манифестация в возрасте 2-15 лет: ригидность, дистония, хореоатетоз, дизартрия, тремор, пирамидные знаки, пигментный ретинит, ночная слепота, амовроз, спастический тетрапарез, слабоумие. При морфологическом исследовании мозга обнаруживают изменения в стриопаллидар-ной системе. Патогенез заболевания неясен. Тип наследования —А/Р.
3. Синдром хореоатетоза, дистонии и паллидарной атрофии. Манифестация в возрасте 5-15 лет: атетоз, торсионный спазм, хореоатетоз, гримасниченье, дизартрия, дисфагия. Смерть наступает спустя 5-10 лет. При патоморфологическом исследовании обнаруживается паплидарная атрофия. Патогенез неясен.
4. Доброкачественная семейная хорея. Манифестация может наступить в раннем или старшем детском возрасте: хорея, нарушения ходьбы, расстройство мимической мускулатуры, дизартрия, интеллект сохранен. Патогенез неизвестен. Тип наследования — А/Д.
5. Семейный непрогрессирующий хореоатетоз. Манифестация заболевания возможна в возрасте 3-8 лет и позже: хореоатетоз без атаксии и тремора, интеллект сохранен. Патогенез неясен.
6. Прогрессирующий атетоз со скелетной дисплазией и кератансульфатурия. Манифестация возможна в возрасте 5-7 лет: атетоз, аномалии ребер и головки бедренной кости, помутнение роговицы, повышенная экскреция кератансульфата с мочой. Умственное развитие в пределах нормы. Предполагается, что в основе заболевания лежит (3-галактозидазная недостаточность. Возможно, речь идет о новой форме мукополисахаридоза.
7. Эпизоды лихорадки с потерей сознания в семьях с атетозом и умственной отсталостью. Заболевание склонны относить к наследственным аминоацидопатиям или другим наследственным метаболическим энцефалопатиям.
- Рекомендуем далее ознакомиться со статьей "Метаболические энцефалопатии детей-подростков: атрофия Corpus Luisi, лейкодистрофии"
Оглавление темы "Метаболические энцефалопатии у детей":- Метаболические энцефалопатии детей ясельного возраста: метахроматическая лейкодистрофия
- Метаболические энцефалопатии детей ясельного возраста: болезнь Аустина, нейроаксональная дегенерация, синдром Луи Бара
- Метаболические энцефалопатии детей ясельного возраста: болезни Нимана-Пика, Дери, Краббе, Гоше
- Метаболические энцефалопатии детей ясельного возраста: нейрональный цероидлипофусциноз, синдром Альперса
- Дифференциация метаболических энцефалопатий детей ясельного возраста
- Метаболические энцефалопатии детей-подростков: болезнь Вильсона-Коновалова, хорея Гентингтона, синдром Ханта
- Метаболические энцефалопатии детей-подростков: прогрессирующая торсионная дистония, болезнь Галлервордена-Шпатца, синдром хореоатетоза
- Метаболические энцефалопатии детей-подростков: атрофия Corpus Luisi, лейкодистрофии
- Метаболические энцефалопатии детей-подростков: атаксия Фридрейха, синдромы Русси-Леви, Ван-Богарта-Шредера-Эпштейна
- Метаболические энцефалопатии детей-подростков: синдром Базена-Корнцвейга, миоклоническая эпилепсия типа Лафора