
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Дифференциация метаболических энцефалопатий детей-подростков
Ранняя диагностика прогрессирующих наследственных энцефалопатий крайне трудна. Это объясняется тем, что дети довольно длительный период развиваются нормально. Первые симптомы настолько неспецифичны, что не позволяют сразу же отнести их к признакам наследственных заболеваний, медленно, но неуклонно прогрессирующих.
Дифференциально-диагностические трудности не уменьшаются с увеличением продолжительности заболевания, так как в клинической симптоматике превалируют тяжелые и сходные психоневрологические расстройства: умственная отсталость, спастический тетрапарез и др. Пытаясь найти для врача более или менее объективные ориентиры, исследователи предлагают из существующих для этих заболеваний симптомокомплексов выделить ряд симптомов.
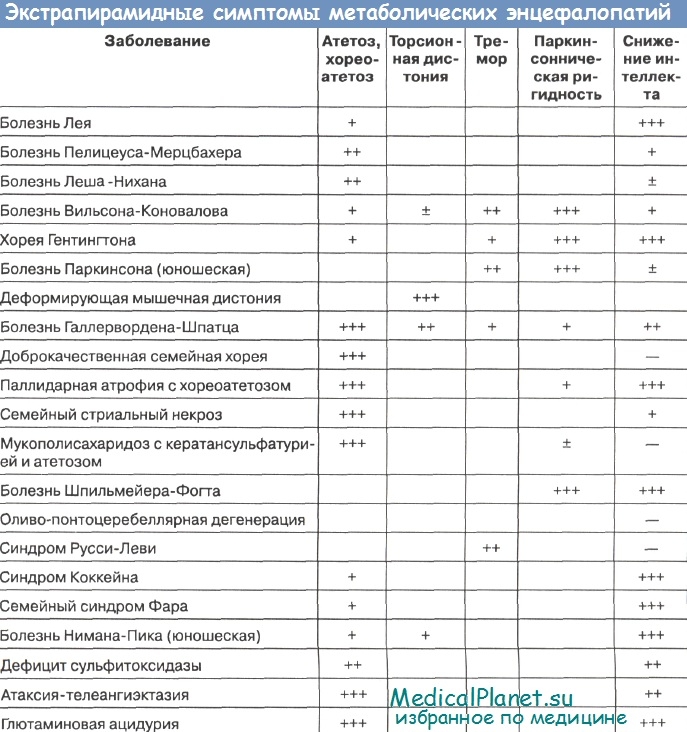
Таким образом, дифференциальный диагноз предлагается осуществлять не только путем сравнения симптомокомплексов разных нозологических форм, но и по наличию отдельных ведущих симптомов: экстрапирамидные знаки, атаксия, судороги, миоклония, полиневриты.

На втором этапе обследования больных этой группы возможности лабораторной диагностики резко ограничены (диагноз базируется только на клинических симптомах и результатах морфологических исследований мозга). Так, окончательный диагноз 13 нозологических форм становится очевидным только при сопоставлении клинических изменений и патоморфологических находок (№№2,3,4,5,6,7, 10,11,14,16, 17, 18, 21).
В диагностике остальных 13 заболеваний могут быть использованы результаты биохимических и морфологических исследований.
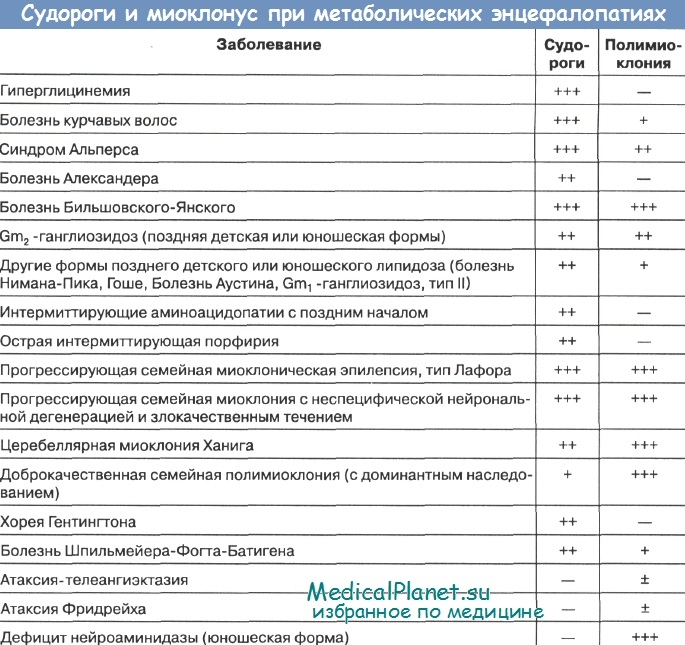
Обращает на себя внимание, что целый ряд заболеваний, при которых выявлен биохимический дефект, не имеют подробной клинической характеристики и рекомендаций по коррекции нарушений метаболизма. Прежде всего, это объясняется их редкостью и небольшим числом наблюдений.
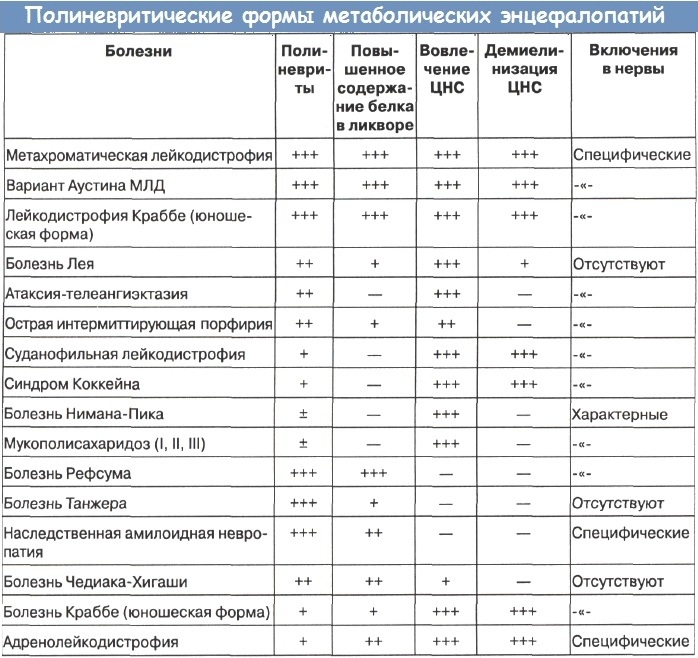
Необходимо отметить, что многие врожденные нарушения обмена в неонатальном периоде протекают весьма остро с явлениями метаболической декомпенсации, ацидозом, рвотой, респираторными расстройствами. Эта симптоматика имитирует часто острый инфекционный процесс и о наследственных заболеваниях обмена даже не возникает подозрений, ни в клинике, ни на секции.

Характер клинических изменений носит смешанный характер. Помимо неврологических расстройств обнаруживается целый комплекс тяжелых изменений различных органов и систем, т.е. заболевания носят полиорганный характер (сочетание первичных и вторично наступающих расстройств). Это позволяет такую патологию рассматривать как раздел нейропедиатрии.

Истинная частота этой группы заболеваний, протекающих остро в неонатальном периоде, остается неясной, так как большая часть из них практически не диагностируется. Исключение могут составлять те патологические состояния, при которых происходит утрата ранее приобретенных способностей.
Редкость большинства нозологических форм и необходимость располагать широкой гаммой биохимических и других, не менее сложных, диагностических методов, не позволяют надеяться на раннюю диагностику. В связи с чем огромное значение, особенно в больших городах, приобретают централизованные диагностические лаборатории, обслуживающие все имеющиеся в городе лечебные учреждения.
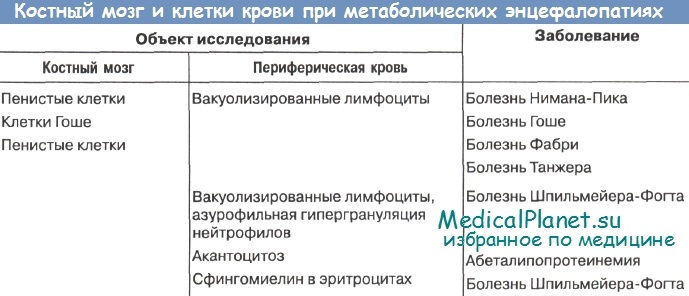
- Рекомендуем далее ознакомиться со статьей "Принципы диагностики наследственных заболеваний по фенотипу"
Оглавление темы "Наследственные болезни у детей":- Метаболические энцефалопатии детей-подростков: болезнь Шпильмейера-Фогта, детская форма болезни Гоше, семейная полимиоклония
- Метаболические энцефалопатии детей-подростков: болезни Рефсума, Танжера, Андерсона Фабри
- Дифференциация метаболических энцефалопатий детей-подростков
- Принципы диагностики наследственных заболеваний по фенотипу
- Cиндром Лоуренса-Муна-Барде-Бидля (ЛМББ) - фенотип, диагностика
- Фенотипы рахитоподобных заболеваний у детей
- Витамин Д-резистентный рахит (ВДРР) - фенотип, диагностика
- Витамин Д-зависимый рахит (ВДЗР) - фенотип, диагностика
- Почечно-канальцевый ацидоз (ПКА) - фенотип, диагностика
- Болезнь де Тони-Дебре-Фанкони (ТДФ) - фенотип, диагностика