
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Болезни с диспропорциональным отставанием роста: мукополисахаридозы
Мукополисахаридозы представляют собой наследственные нарушения обмена гликозаминогликанов и относятся к болезням накопления - лизосомальным болезням. Ряд типов мукополисахаридозов клинически характеризуется системным поражением опорно-двигательной системы. В данную дифференциально-диагностическую группу включены те из них, при которых отчетливо выражены нарушения роста.
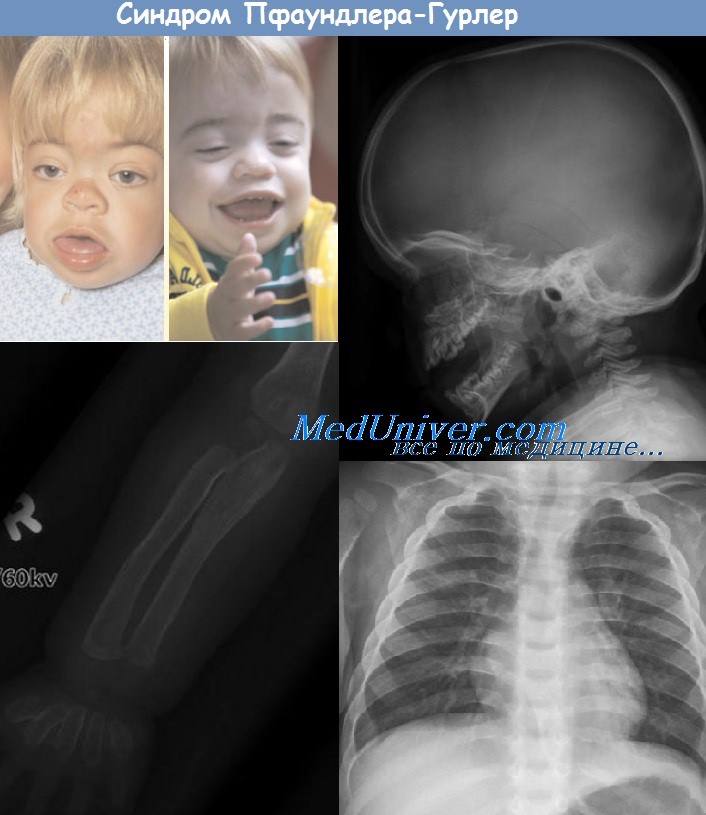
Мукополисахаридоз I-II типа (синдром Гурлер) описан в 1919 г. Hurler. Обусловлен дефектом фермента a-L-идуронидазы. Клинически проявляется с первых месяцев жизни. Резкие деформации скелета и черепа. Грубые черты лица. Гипертелоризм. Эпикант. Широкий нос с приплюснутой переносицей и вывернутыми ноздрями. Большие и толстые губы.
Часто открытый рот, большой язык. Маленькие, широко расставленные зубы. Хронический ринит. Резкое отставание в росте. Короткая шея.
Кифоз с горбом в нижнем грудном и верхнем поясничном отделе. Большой живот. Гепатоспленомегалия. Широкие кисти с короткими пальцами. Сгибательная контрактура. Умственная отсталость. Паховые и пупочные грыжи. Гирсутизм. Помутнение роговицы. Глухота. Rg - кубовидные тела позвонков. Кифоз. Утолщение ключиц, лопаток. Деформация тазового кольца.
Уплощение, уменьшение головок бедер. Запаздывание формирования ядер окостенения. Выраженные деформации лицевых костей. Увеличена экскреция с мочой дерматансульфата и гепарансульфата. Тип наследования - А/Р.
Мукополисахаридоз II типа (синдром Гунтера) описан Hunter в 1917 г. Обусловлен дефицитом идуронатсульфатазы. Клинические признаки появляются на 2-4-м году жизни: отставание в росте, умеренные костные деформации, грубые черты лица, гипертелоризм, плоская переносица с большими ноздрями, толстые губы, макроглоссия, широко расставленные зубы, короткая шея. Встречаются контрактуры суставов.
Глухота. Умственная отсталость. Гепатоспленомегалия. Грыжи. Гипертрихоз. Rg -изменения, аналогичные синдрому Гурлер, но менее выражены. Увеличена экскреция с мочой дерматансульфата и гепарансульфата. Тип наследования - рецессивный, сцепленный с Х-хромосомой.
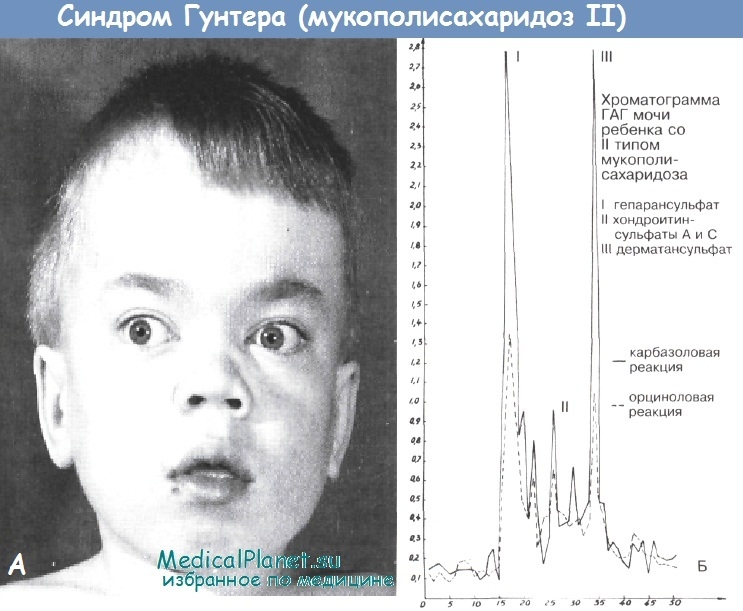
Б - Хроматограммы кислых гликозаминстликанов.
Мукополисахаридоз IV типа (синдром Моркио) описан в 1929 г. Morquio. Обусловлен дефицитом фермента хондроитин-6-сульфат-ацетилглюкозамин-4-сульфат-сульфатазы. Клиническая симптоматика проявляется на 1-3 годах жизни: отставание в росте, значительные деформации скелета (особенно грудной клетки), широкий рот, выступающая верхняя челюсть, короткий нос, широко расставленные зубы, короткая шея, кифоз, резкая килевидная деформация грудной клетки.
Интеллект нормальный. Движения в суставах верхних конечностей ограничены. Наблюдаются вальгусная деформация голеней и стоп, помутнение роговицы, тугоухость, склонность к простудным заболеваниям, грыжа, гепатомегалия, кардиопатии (иногда). Rg - платиспондилия. Выраженный остеопороз, сколиоз. Гипоплазия зубов. Расширение метафизов. Головки бедер уплощены, фрагментированы. Запаздывание появления ядер окостенения запястий. Конусовидное сближение проксимальных концов пястных костей.
Увеличена экскреция с мочой кератансульфата. Тип наследования - А/Р. В последние годы MPS-SB связывают с р-галактозидазной недостаточностью.

Мукополисахаридоз V типа (синдром Шейе). Клиническая картина складывается из наличия грубых внешних черт, тугоподвижности суставов и помутнения роговицы. Рост больных может быть не изменен или несколько снижен. Тип наследования - А/Р. При определении содержания ГАГ в моче обнаруживаются их значительное увеличение, в основном за счет дерматансульфата. При изучении биоптатов и функции печени выявляются грубые нарушения ее структуры и функции.
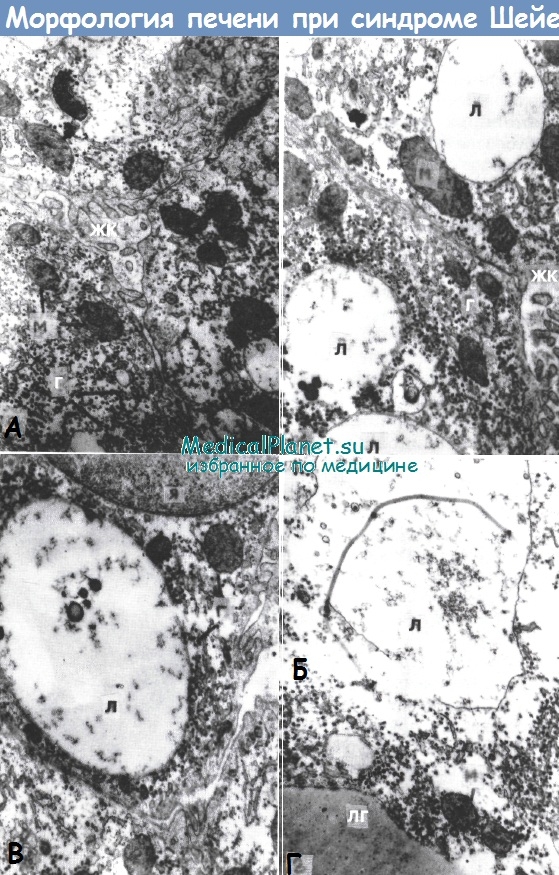
Грубые нарушения структуры и функции печени.
А, Б, В, Г (х7600) - последовательные стадии дистрофических нарушений гепатоцитов, связанных с увеличением «вакуолей накопления»,
Мукополисахаридоз VI типа (синдром Марото-Лами) описан в 1963 г. Maroteaux и Lamy. Обусловлен дефектом фермента арилсульфатазы В. Первые клинические признаки появляются на 1-3-м годах жизни: резкое отставание в росте, макроцефалия, грубое лицо, гипертелоризм, большой нос, толстые губы, макроглоссия, короткая шея, бочкообразная грудная клетка, кифоз (иногда).
Наблюдаются сгибательные контрактуры в суставах; вальгусная деформация голеней; помутнение роговицы вплоть до слепоты; глухота (иногда); паховые, пупочные грыжи; гепатоспленомегалия; Rg - деформация тазового кольца; истончение шейки бедер; округлая двояковыпуклая форма позвонков; вогнутая задняя поверхность поясничных позвонков. Интеллект не изменен. Увеличена экскреция с мочой дерматансульфата. Тип наследования - А/Р.

- Рекомендуем далее ознакомиться со статьей "Дифференциация заболеваний с пропорциональным нарушением роста"
Оглавление темы "Врожденные болезни опорно-двигательного аппарата":- Наследственные болезни опорно-двигательной системы - типы наследования
- Дифференциация заболеваний с диспропорциональным нарушением роста
- Болезни с диспропорциональным отставанием роста: ахондроплазия, гипохондроплазия, дисплазии
- Болезни с диспропорциональным отставанием роста: метафизарная хондродисплазия Шмидта, несовершенное костеобразование, синдром Блюма
- Болезни с диспропорциональным отставанием роста: ухо-небно-пальцевой синдром, синдром Вейля-Маркезани, болезнь Эллиса-ван-Кревельда
- Болезни с диспропорциональным отставанием роста: мукополисахаридозы
- Дифференциация заболеваний с пропорциональным нарушением роста
- Болезни с пропорциональным отставанием роста: гипофизарная карликовость, синдромы Корнелия де Ланге и Секеля
- Болезни с пропорциональным отставанием роста: синдромы Рассела-Сильвера, Дубовича, Рубинштейна-Тейби
- Болезни с пропорциональным отставанием роста: лепречаунизм, синдромы Смита-Лемли-Опитца, Нунана, Ханхарта, болезнь Альберса-Шенберга