
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
III тип гликогеноза (болезнь Кори) - причины, клиника
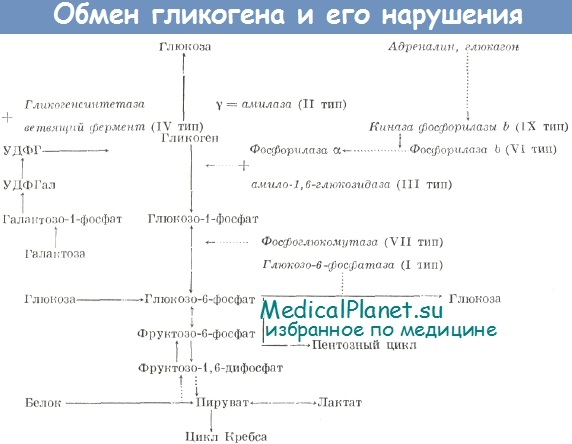
III тип гликогеноза (болезнь Кори) вызван полным отсутствием или снижением активности амило-1,6-глюкозидазы — фермента, расщепляющего а-1,6-глюкозидные связи в точках ветвления гликогена. Этот фермент вместе с фосфорилазой способствует полному распаду гликогена. Заболевание наследуется по аутосомно-рецессивному типу.
Известно, что фосфорилаза расщепляет наружные ветви молекулы гликогена, оставляя при этом по 3—4 глюкозных остатка. Дальнейшее ферментативное расщепление гликогена осуществляется в два этапа:
1) перенос двух или трех глюкозных остатков с цепи А на цепь В молекулы гликогена (олиго-1,4-1,4-трансглюкозилазная активность) и
2) собственно отщепление остатка глюкозы, связанного с цепью В а-1,6-глюкозидной связью (амило-1,6-глюкозидазная активность). Лимитирующей реакцией является гидролиз а-1,6-глюкозидных связей.
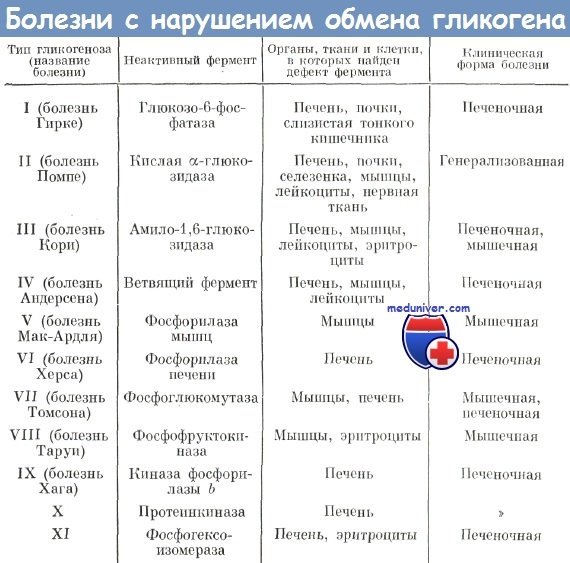
Больные с III типом гликогеноза могут быть отнесены к различным подтипам в зависимости от того, отсутствуют ли обе активности фермента или одна из них, а также от того, в каких органах и тканях отсутствует ферментативная активность.
Van Hoof и Hers предложили следующую классификацию III типа гликогеноза.
По клинической картине подтипы А, В и D III типа гликогеноза относят к печеночной форме заболевания.
В печени накапливается аномальный по структуре гликоген, «инертный» для обмена. Глюкоза, поступающая в клетки печени, может участвовать в создании наружных ветвей молекулы гликогена. Эта возможность, однако, ограничена, и поэтому печень поддерживает гомеостаз глюкозы крови в течение нескольких часов. Нарушение в распаде гликогена сопровождается активацией компенсаторных механизмов, способствующих нормализации содержания глюкозы в крови.
Усиливается распад белков, при этом часть аминокислот используется для образования глюкозы (глюкогениые аминокислоты), а часть аминокислот образует ацетил-КоА (кетогенные аминокислоты). Нарушаются ферментативные процессы цикла Кребса, что приводит к избыточному синтезу ацетона и кетоновых тел. У больных происходит усиленный распад жиров натощак, вызванный гипогликемией, что также способствует образованию в организме избытка кетоновых тел и ацетона.
Активность гликогенсинтетазы снижена из-за накопления значительных количеств гликогена в гепатоцитах. Поступающая экзогенным путем глюкоза используется для синтеза жиров, а не для синтеза гликогена.
Клиническая картина заболевания характеризуется гипогликемией и ацетонемией натощак, а также характерным внешним видом детей.
При анализе крови обнаруживают умеренно выраженную нормохромную анемию, повышенное содержание липидов, ацетона и кетоновых тел. Концентрация лактата в крови соответствует норме.
При анализе мочи отклонений от нормы не наблюдают.
Гликемические кривые в ответ па пероральное введение глюкозы необычны и имеют двух- или трехгорбный вид, что, по всей видимости, характерно для этого типа заболевания. Содержание НЭЖК в крови падает сразу после приема глюкозы, а к концу 3-го часа вновь повышается.
При введении галактозы, по данным ряда авторов, происходит увеличение концентрации глюкозы и лактата в крови. Такой же характер гликемических и лактатемических кривых наблюдается и при введении фруктозы.
Результаты, полученные при проведении адреналиновой и глюкагоновой нагрузок, зависят от того, через какой промежуток времени после еды проводится нагрузка. Так, например, если проводить исследование через 12—14 ч после еды, то за такой срок наружные ветви гликогена под действием фосфорилазы укорачиваются и образуется фосфорилазпый конечный декстрин, который не может использоваться клетками печени, и введение адреналина или глюкагона не вызывает подъема содержания глюкозы в крови.
Если же нагрузочную пробу с адреналином или глюкагоном проводить у больных с III типом гликогеноза через 3—5 ч после еды, когда успевают образоваться более длинные наружные ветви гликогена, то наблюдается такое же, как в норме, увеличение концентрации глюкозы в крови.
Так как глюкогеногенез у больных болезнью Кори не нарушен, при нагрузочной пробе с пероральным введением аминокислот или белка наблюдается повышение концентрации глюкозы в крови, которое может сохраняться на протяжении 3—4 ч (Fernandes, Van de Kamer).
Для окончательного диагноза заболевания необходимо проведение биохимического исследования пораженной ткани больного. Исследуется как общая активность фермента, так и отдельно его трансглюкозилазная и гидролитическая активности. Для установления подтипа заболевания необходимо определение обеих ферментативных активностей во всех органах и тканях больного.
При III типе гликогеноза наиболее часто наблюдается дефект активности как амило-1,6-глюкозидазы, так и глюкозо-6-фосфатазы, однако это не двойной генетический дефект, так как подавление глюкозо-6-фосфатазной активности вторично. В некоторых случаях мы наблюдали также снижение активности фосфорилазы, что усложняло диагноз заболевания и утяжеляло течение болезни.
Важным в диагностике гликогеноза III типа является определение концентрации и структуры полисахарида в органах и тканях больного.
С помощью различных методов исследования удалось выяснить, что у больных с дефицитом амило-1,6-глюкозидазы накапливается в клетках большое количество фосфорилированного декстрина гликогена (лимитдекстрина), который не вступает в обменные реакции.
Иногда активность амило-1,6-глюкозидазы отсутствует не только в гепатоцитах, но и в лейкоцитах, и эритроцитах, что приводит к накоплению в них лимитдекстрина. Факт отсутствия активности амило-1,6-глюкозидазы в форменных элементах крови широко используется в диагностике заболевания. В ряде случаев гликогеноза III типа отсутствие активности амило-1,6-глюкозидазы в печени не сопровождается отсутствием активности фермента и накоплением аномального по структуре гликогена в эритроцитах и лейкоцитах.
В культуре фибробластов кожи больных с III типом гликогеновой болезни активность амило-1,6-глюкозидазы в некоторых случаях может отсутствовать, что дает основание считать возможной пренатальную диагностику III типа гликогеноза.
При гистохимическом исследовании гепатоцитов находят их жировую инфильтрацию и накопление полисахарида в цитоплазме. При электронно-микроскопическом исследовании гепатоцитов полисахарид находится в цитоплазме в виде b-частиц. В мышцах значительное накопление полисахарида имеется в саркоплазме, между миофибриллами, а также в виде р-частиц.
Прогноз печеночной формы III типа гликогеновой болезни, как правило, благоприятен. Данное заболевание наиболее опасно в возрасте 4—5 лет, когда часты приступы гипогликемии, сопровождающиеся метаболическим ацидозом и ацетонемией. В более зрелом возрасте симптомы заболевания сглаживаются.
- Рекомендуем далее ознакомиться со статьей "IV тип гликогеноза (болезнь Андерсена) - причины, клиника"
Оглавление темы "Нарушения обмена углеводов у детей":- Гликогеновая болезнь (гликогеноз) - клиника, проявления
- I тип гликогеноза (болезнь Гирке) - причины, клиника
- II тип гликогеноза (болезнь Помпе) - причины, клиника
- III тип гликогеноза (болезнь Кори) - причины, клиника
- IV тип гликогеноза (болезнь Андерсена) - причины, клиника
- V тип гликогеноза (болезнь Мак-Ардля) - причины, клиника
- VI тип гликогеноза (болезнь Херса) - причины, клиника
- VII, VIII типы гликогеноза (болезни Томсона, Таруи) - причины, клиника
- IX тип гликогеноза (болезнь Хага) - причины, клиника
- Методы диагностики гликогенозов (гликогеновой болезни) - анализы