
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Синдром Хатчинсона-Гилфорда или прогерия детей. Синдром Вернера.
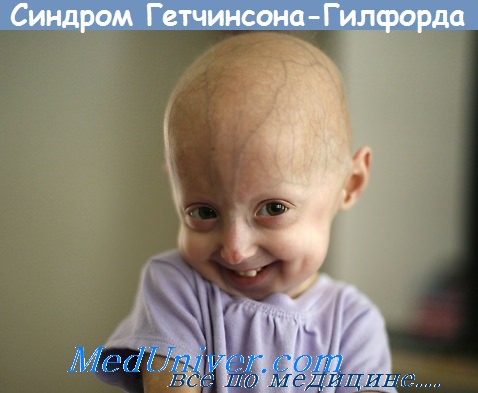
Синдром Хатчинсона-Гилфорда, или прогерия детей, - крайне редкое заболевание. Его частота составляет 1 на 1 000 000 человек. Именно этот синдром занесен под названием прогерии в OMIM (Online Mendeltan inheritance in Man) - электронную версию каталога В. Маккьюсика, атакже в POSSUM и Oxford Medical Database - ведущие компьютерные базы данных по наследственной патологии. Фенотип пациентов чрезвычайно характерный: маленький рост, «птичье» лицо с клювообразным профилем, преобладание размеров мозговой части черепа над лицевой, выступающая венозная сеть на коже мозговой части, как правило, обнаженной вследствие аллопеции, часто тотальной, с выпадением бровей и ресниц. Наблюдается резкая гипоплазия ключиц, дефекты формы/числа зубов, сухая истонченная кожа, практически полное отсутствие подкожной жировой клетчатки, отставание в развитии, особенно физическом. Больные бесплодны, хотя в литературе описан случай рождения ребенка у пацентки с синдромом Хатчинсона-Гилфорда. В крови повышен уровень Средняя продолжительность жизни описанных носителей синдрома- 13,4 лет (как редкое наблюдение описан единственный 45-летний пациент). Причиной смерти, как правило, служит инфаркт миокарда, с выявлением на аутопсии генерализованного атеросклероза и фиброза миокарда, а также отложения жироподобного вещества в тканях мозга и паренхиматозных органов.
Репарация ДНК при синдроме Хатчинсона-Гилфорда нарушена: установлено, что клетки его носителей не способны избавляться от вызываемых химическими агентами сшивок ДНК-белок. Но главная диагностическая особенность клеток больных с данным синдромом состоит в резко сниженном, по сравнению с нормой, количестве делений, которое способны пройти клетки в культуре (так называемый лимит, или число Хейфлика). В 1971 г. А.М. Оловников высказал предположение об укорочении хромосомных теломер в процессе развития клеток. А в 1992 г. было показано, что для клеток пациентов с синдромом Хатчинсона-Гилфорда характерно врожденное укорочение теломер. Анализ взаимосвязи между лимитом Хейфлика, длиной теломер и активностью теломеразы (фермента, способного наращивать 3'-конец теломерной ДНК) дает возможность соотнести естественное старение и процесс формирования клинической картины при синдроме Хатчинсона-Гилфорда,
Крайне низкая частота встречаемости данной формы прогерии позволяет лишь высказывать гипотезы о типе наследования. Аутосомно-рецессивный тип предполагается по аналогии с синдромом Коккейна, имеющим отдельные черты преждевременного старения. Но есть и предположение о развитии синдрома Хатчинсона-Гилфорда вследствие доминантной аутосомной мутации, возникшей de novo. Оно получило косвенное подтверждение на основе измерения теломер у носителей синдрома, их родителей и здоровых доноров. Было показано, существенное укорочение тело-мер у больных по сравнению с другими двумя испытуемыми группами.
Синдром Вернера
Синдром Вернера (WS), или прогерия взрослых, также характеризуется симптомами преждевременного старения, но возникающими уже после завершения полового созревания. Пациенты седеют и теряют волосы еще в возрасте до 20 лет. Появляются старческие изменения кожи (морщины, гиперпигментация, сухость, гиперкератоз, телеангиэктазии), голос утрачивает звонкость. Кроме того, наблюдается широкий спектр патологии, сопровождающей обычное старение: атеросклероз, остеопо-роз, катаракта, диабет, различные типы доброкачественных и злокачественных опухолей. Отмеченный высокий уровень гиалуроновой кислоты в моче совпадает с таковым и в случае вышеописанной прогерии Хатчинсона—Гилфорда. Аналогичная картина наблюдается и в отношении лимита Хейфлика. Клетки больных WS исчерпывают свой пролиферативный потенциал в 3-5 раз быстрее, чем у здоровых доноров, но растут значительно медленнее нормальных (заметим, что продолжительность клеточного цикла в случае прогерии Хатчинсона—Гилфорда соответствует норме). Отсутствие врожденного феномена укороченных теломер у больных WS — еще один признак, по которому разнятся эти две формы прогерии. Вследствие этого уменьшенный лимит Хейфлика у больных WS связывают с нарушением регуляции остановки клеточных делений при укорочении теломер до определенного уровня. Хотя WS и является достаточно редко встречающимся аутосомно-рецессивным заболеванием, его частота в Японии составляет 1 на 40 000 человек, что на два порядка выше, чем для синдрома Хатчинсона-Гилфорда. Ген, ответственный за прогерию взрослых и названный WRN, картирован в локусе (р12-р21) хромосомы 8. Продуцируемый им белок WRN выполняет функцию геликазы, но в отличие от ХРВ, XPD и CSB его активность, по-видимому, не вовлечена в процесс репарации ДНК, связанный с транскрипцией: WRN не входит в основной транскрипционный комплекс человека TFIIH.
Многие из патологических симптомов синдрома Вернера сходны с теми, что наблюдаются в процессе нормального старения, но есть и отличия. Так соотношение количества эпителиальных и неэпителиальных опухолей у пациентов с WS составляет 1:1, а в обшей популяции - 10:1. Кроме того, по сравнению с обшей популяцией, реже наблюдаются гипертензия и поражения центральной нервной системы.
Синдром Вернера и описанные выше пигментная ксеродерма, анемия Фанкони, атаксия-телеангиэктазия и синдром Блума объединены общим признаком — генетически детерминированной нестабильностью хромосом. Этот феномен выражается в постоянном, возникающем спонтанно или под действием некоторых агентов, изменении структуры хромосом, их отдельных локусов или групп локусов. Результатом являются мутации соматических клеток и злокачественные новообразования.
Отдельные нарушения работы систем репарации имеют место и при более распространенных заболеваниях - системных (ревматоидный артрит, системная красная волчанка и др.), некоторых нейродегенеративных (например, при болезни Альцгеймера) и онкологических заболеваниях.
В 1994 г, было показано, что мутации в гене MSH2 у человека, сходном с аналогичными генами у S. cerevisae и геном mutS у Е. coti, приводят к нарушению репарации одного из типов репликативных ошибок - неправильно спаренных оснований вследствие небольших делеций (утрат) или инсерций (вставок). Мутация водной из копий этого гена с высокой вероятностью детерминирует развитие некоторых видов новообразований: семейного неполипозногорака кишечника (HNPCC) и рака эндометрия. В опухолевых клетках потеряна вторая копия гена, и наблюдается высокая мутабильность микросателлитных повторов (возрастающая, по крайней мере, в 100 раз). По аналогии были найдены еще несколько генов, один из которых, MLH1 гомологичен mutL гену у бактерий. Мутации в этом гене также приводят к развитию неполипозного рака кишечника. Таким образом, существует принципиальное сходство некоторых систем репарации у про- и эукариот.
- Читать далее "Комбинативная изменчивость. Генетическая рекомбинация."
Оглавление темы "Рекомбинация и транскрипция ДНК.":1. Атаксия-телеангиэктазия или синдром Луи-Бар. Синдром Блума.
2. Синдром Хатчинсона-Гилфорда или прогерия детей. Синдром Вернера.
3. Комбинативная изменчивость. Генетическая рекомбинация.
4. Модель Холлидея. Структура Холлидея.
5. Модель Мезельсона-Реддинга. Модель Жостака в генетике.
6. Генная конверсия. Механизмы генной конверсии.
7. Сайт специфическая рекомбинация. Особенности сайт специфической рекомбинации.
8. Незаконная рекомбинация в генетике. Методика незаконной рекомбинации.
9. Регуляция генной активности в ДНК. Транскрипция и регуляция генной активности.
10. Этапы транскрипции. Особенности транскрипции.