
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Атаксия-телеангиэктазия или синдром Луи-Бар. Синдром Блума.
Изучение эксцизионной репарации, проводившееся в течение последних 40 лет, показало, что у эукариот в целом она организована сходным образом. Описаны многие гены, ответственные за синтез и работу ферментов репарации ДНК после УФ-облучения. Мутации в этих генах приводят к развитию патологических признаков при целом ряде наследсгвенных заболеваний, сопровождающихся снижением средней продолжительности жизни. Нарушения в работе репаративных ферментов значительно увеличивают риск развития онкологических заболеваний у человека.
Механизмы генетической репарации после действия ионизирующей радиации изучены значительно хуже, в первую очередь, вследствие большого разнообразия типов индуцированных радиацией повреждений ДНК. Однако значительная часть этих повреждений устраняется с помощью системы эксцизионной репарации. Но в ликвидации двухцепочечных разрывов используются другие механизмы, а именно -мейотической рекомбинации. В настоящее время возможности изучения таких механизмов у человека практически ограничены исследованиями клеток больных атаксией-телеангиэктазией.
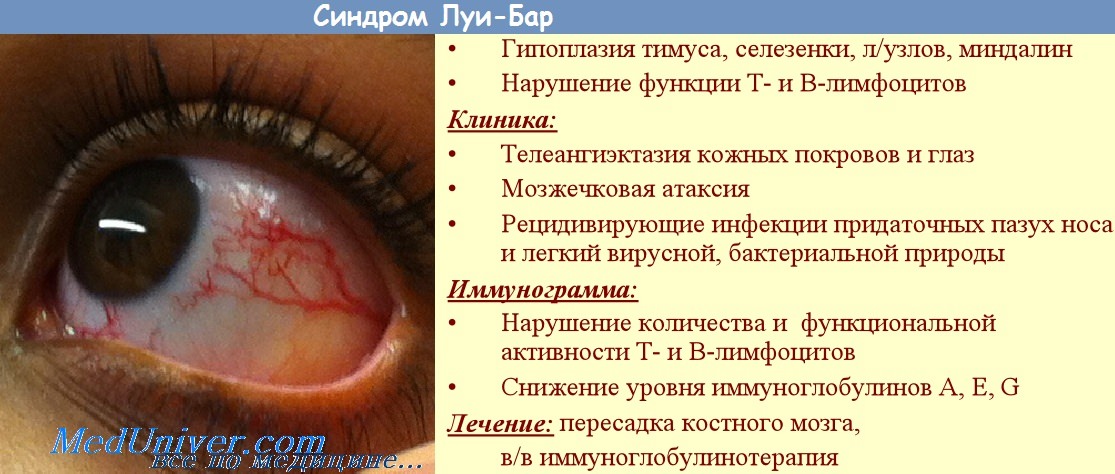
Атаксия-телеангиэктазия (AT), или синдром Луи-Бар, — аутосомно-рецессивное заболевание, характеризующееся мозжечковой атаксией (расстройством координации движений), телеангиэктазами (локальным чрезмерным расширением мелких сосудов), различными формами иммуннодефицита, предрасположенностью к онкологическим заболеваниям. Частота синдрома составляет от 1/100 000 до 1/40 000 человек. Пациенты, как правило, не доживают до 50 лет.
Немаловажно, что при AT наблюдается повышенная частота спонтанных и индуцированных хромосомных перестроек в соматических клетках, а также повышенный уровень общей хромосомной нестабильности в лимфоцитах. Хромосомы этих больных имеют значительно укороченные теломеры (примерно втрое по сравнению с теломерами здоровых доноров). В 1995 г. П. Гринвелл установил, что ген TEL1 Saccharomyces cerevisiae, ответственный за укорочение теломер у дрожжей, является гомологом гена AT— atm (AT-mutated). Клетки всех без исключения изученных до сих пор больных AT аномально чувствительны к воздействию ионизирующей радиации и радиомиметиков (химических веществ, имитирующих действие ионизирующего излучения). Это проявляется в пониженной выживаемости клеток и повышенном уровне хромосомных аномалий.
Характерная особенность соматических клеток больных AT — радиорезистентность синтеза ДНК. Последнее свойство очень важно, и помогает понять молекулярный механизм возникновения болезни. В норме в клеточном цикле существуют две точки задержки клеточного деления в случае возникновения значительных повреждений ДНК: G1-S и G2-M. Эти остановки необходимы для исправления повреждений ДНК. У больных атаксией-телеангиэктазией клетки не останавливаются перед фазой синтеза ДНК и, следовательно, у них нет времени для рспарирования повреждений ДНК. Как полагает ряд исследователей, одна из причин данного феномена кроется в том, что ключевой белок Р53, в норме отвечающий за упомянутую выше остановку, не индуцируется при наличии повреждений.
Синдром Блума
Синдром Блума (пропорциональная пре- и постнатальная задержка роста, чувствительность к ультрафиолету солнечных лучей, гипо- и гиперпигментироаанная кожа, характерная краснота на лице в виде бабочки, предрасположенность к онкологическим заболеваниям и хромосомная нестабильность) также наследуется по аутосомно-рецессивному типу Клетки больных с синдромом Блума характеризуются высоким уровнем спонтанных хромосомных аберраций и сестринских хроматидных обменов (СХО). Заболевание обусловлено мутациями в гене BLM (Bloom-mutated), кодирующем белок, сходный с RecQ-геликазой Е. coli. Этотбелок у Е. соli является участником RecF-рекомбинационногопути. Ген BLM картирован на хромосоме 15 в лоryce q26.1, в той же области, где локализован протоонкоген fes. Предполагается, что ген имеет ДНК-зависимую АТРазную активность и ДНК-геликазную активность. С негеликазной частью исследователи связывают функцию поддержания стабильности хромосом в клетках, а ДН К-геликазной части белка BLM отводят ключевую роль на некоторых этапах репарации.
Известны и другие нозологические формы, клинические проявления которых тоже связывают с дефектами репарации ДНК, но уже безотносительно ионизирующего или УФ-облучения. К таким заболеваниям относятся анемия Фанкони и прогерии - синдромы Вернера и Хатчинсона-Гилфорда.
В случае анемии Фанкони (FA), наследующейся по аутосомно-рецессивному типу и характеризующейся поражением всех элементов костного мозга (и как следствие — снижением количества всех клеточных элементов крови), наблюдается нарушение вырезания пиримидиновыхдимеров, а также нарушение репарации межцепочечных сшивок ДНК У больных FA и их кровных родственников установлена повышенная частота новообразований. Частота острой миелоидной лейкемии в у FA-пациентов превышает популяционную среднюю в 15000 раз. Клетки этих больных характеризуются сниженной выживаемостью после воздействия химических агентов, вызывающих поперечные сшивки цепей ДНК, и отсутствием такого эффекта в ответ на действие у-УФ-лучей. Кроме того, фаза G2 в FA-клетках продолжается в 2 раза дольше, чем в клетках здоровых доноров, что до сих пор не получило объяснения. Для FA выделено 8 групп комплементации (А-Н), из которых для двух идентифицированы гены- FAA и FAC (ген картирован на длинном плече хромосомы 9 в лoкyce q22.3).
Возможно, белковые продукты этих генов представляют собой часть сложной репарационой системы, но сам дефект репарации не является первичным для сложного фенотипа FA.
Прогерии - это заболевания, основным нозологическим признаком которых является преждевременное старение. По отношению к началу процесса старения (до или после полового созревания) выделяют две основные формы -синдром Хатчинсона-Гилфорда и синдром Вернера.
- Читать далее "Синдром Хатчинсона-Гилфорда или прогерия детей. Синдром Вернера."
Оглавление темы "Рекомбинация и транскрипция ДНК.":1. Атаксия-телеангиэктазия или синдром Луи-Бар. Синдром Блума.
2. Синдром Хатчинсона-Гилфорда или прогерия детей. Синдром Вернера.
3. Комбинативная изменчивость. Генетическая рекомбинация.
4. Модель Холлидея. Структура Холлидея.
5. Модель Мезельсона-Реддинга. Модель Жостака в генетике.
6. Генная конверсия. Механизмы генной конверсии.
7. Сайт специфическая рекомбинация. Особенности сайт специфической рекомбинации.
8. Незаконная рекомбинация в генетике. Методика незаконной рекомбинации.
9. Регуляция генной активности в ДНК. Транскрипция и регуляция генной активности.
10. Этапы транскрипции. Особенности транскрипции.