
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Фенилкетонурия. Признаки и причины фенилкетонурии.
Фенилкетонурия (фенилаланинкетонурия, фенилпировиноградная олигофрения, ФКУ) - заболевание, обусловленное недостаточной (отсутствием или снижением) активностью фенилаланингидроксилазы. Первое описание этого заболевания принадлежит Фёллингу, который в 1934 г. обнаружил, что моча двух сибсов с умственной отсталостью при добавлении к ней треххлорного железа окрашивается в зеленый цвет. Кроме того Фёллинг доказал наследственный характер заболевания. Впоследствии Пенроуз установил аутосомно-рецессивный характер заболевания. Средняя частота ФКУ составляет 1:10000 новорожденных, а наибольшая распространенность характерна для популяций Ирландии и Шотландии, где ее частота достигает 1:4500 новорожденных, а также Турции — 1:2600. В нашей стране распространенность ФКУ не превышает 1: 10000 новорожденных.
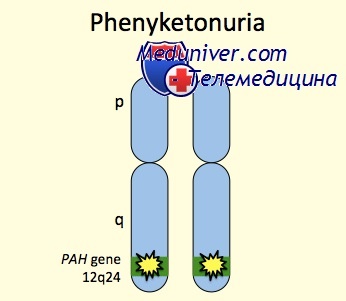
Ген, кодирующий ФАГ, локализован в длинном плече 12-й хромосомы (12q22-q24.2). Основной тип мутаций в гене, обозначаемом РАН, - это однонуклеотидные замены, которые в большинстве случаев локализованы в 7, 9 и 12 экзонах. Показано, что в различных странах Европы и Азии встречается определенный спектр мутаций, одна из которых - мажорная. В России и странах Восточной Европы мажорной является однонуклеотидная замена в 12 экзоне, приводящая к замене аргинина на триптофан (R408W). С ней связаны 70% случаев. В Японии и Китае данная мутация не встречается. Эти данные свидетельствуют о том, что замена аргинин-триплета на триптофан-триплет произошла уже после разделения рас.
В основе патогенеза ФКУ - накопление фенилаланина и продуктов побочных реакций - фенилпировиноградной, фенилуксусной ифенилмолочной кислот, а также фенилэтиламина и фенилацетилглугамина, оказывающих токсическое действие на мозг и клетки внутренних органов больного. Ферментативный блок превращения фенилаланина в тирозин приводит также к уменьшению образования медиаторов центральной нервной системы (дофамина и диоксифенилаланина), что вносит вклад в развитие симптомов поражения ЦНС. Кроме этого наблюдается дефицит одного из конечных продуктов обмена фенилаланина - меланина, обусловливающий снижение пигментации кожи, волос, радужной оболочки глаз (дети голубоглазые и светловолосые, со светлой кожей).
Клиническая картина заболевания развивается уже спустя 2-3 недели после рождения. Первыми симптомами могут быть повышенная возбудимость, гиперрефлексия и мышечный гипертонус, тремор. Моча и пот таких больных имеют специфический запах, который иногда ассоциируют с «мышиным», из-за накопления фенилпировиноградной кислоты (ФПВК) и других метаболитов фенилаланина. Клинические проявления по мере накопления фенилаланина и его дериватов нарастают, и к шестимесячному возрасту у ребенка формируются необратимые изменения ЦНС и других органов и систем (дети с ФКУ склонны к дерматитам). Для больных ФКУ характерна прогрессирующая умственная отсталость, эписиндром (причем у 40% больных наблюдаются эпиприступы, а у 85—90% больных регистрируют на ЭЭГ эпилептическую активность) и другие психоневрологические расстройства. В тяжельк случаях моторное развитие практически отсутствует.
Причиной атипичной ФКУ или ФКУ с легким течением может быть гетерозиготность по мутациям, полученным больным ребенком от родителей, так как для обеспечения нормального физического и умственного развития без элиминанионной диеты достаточна ФАГ с 10%-ой активностью от нормы.
- Читать далее "ФАГ независимые гиперфенилаланинемии. Транзиторные формы гиперфенилаланинемии."
Оглавление темы "Наследственные болезни.":1. Синдром Прадера-Вилли. Характеристика синдрома Прадера-Вилли.
2. Синдром Энгельмана. Болезнь экспансии тринуклеотидных повторов.
3. Хорея Гентингтона. Признаки и причины хореи Гентингтона.
4. Миотоническая дистрофия Куршмана-Штейнерта-Баттена. Признаки и причины миотонической дистрофии Куршмана-Штейнерта-Баттена.
5. Синдром ломкой Х хромосомы. Синдром Мартина-Белл.
6. Прионные болезни. Признаки прионных болезней.
7. Наследственные болезни обмена. Классификация и признаки наследственных болезней обмена.
8. Гиперфенилаланинемии. Признаки и причины гиперфенилаланинемий.
9. Фенилкетонурия. Признаки и причины фенилкетонурии.
10. ФАГ независимые гиперфенилаланинемии. Транзиторные формы гиперфенилаланинемии.