
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) синдрома Альпорта
Наследственный нефрит — это гетерогенная группа семейных почечных заболеваний, ассоциированных прежде всего с повреждением клубочков. Особого внимания заслуживают два заболевания этой группы: синдром Альпорта, т.к. хорошо изучены его характерные генетические нарушения, и болезнь тонких базальных мембран, которая проявляется семейной доброкачественной гематурией.
Полная клиническая картина при синдроме Альпорта: гематурия с последующей хронической почечной недостаточностью, нейрогенная глухота и различная патология глаз, в т.ч. дислокация хрусталика, дистрофия роговицы и задняя катаракта.
В 85% наблюдений заболевание сцеплено с Х-хромосомой: у мальчиков развивается полная клиническая картина, а у девочек, являющихся носителями дефектного гена, заболевание ограничивается гематурией. Существуют также аутосомно-доминантные и аутосомно-рецессивные типы наследования синдрома Альпорта, при которых развитие полной клинической картины от пола не зависит.
а) Патогенез. Симптомы заболевания обусловлены нарушением аз-цепи (COL4A3), а4-цепи (COL4A4) или а5-цепи (COL4A5) коллагена типа IV. Классический синдром Альпорта (сцепленный с Х-хромосомой) вызван мутацией COL4A5, а аутосомные варианты — мутациями COL4A3 и COL4A4. Во всех случаях нарушается синтез коллагена типа IV, являющегося основным белком для функционирования БМК, хрусталика и улитки.
Поскольку БМК представляет собой сеть тримерных молекул коллагена из а3-, а4- и а5-цепей, мутация COL4A5 нарушает процесс формирования коллагеновой сети. Поскольку аз-цепь включает антиген Гудпасчера, то клубочки у пациентов с синдромом Альпорта без этой цепи не реагируют с антителами к БМК, взятыми от пациентов с синдромом Гудпасчера.
б) Морфология. При гистологическом исследовании видно, что клубочки вовлечены в патологический процесс. Ранние стадии заболевания можно выявить только при электронной микроскопии по диффузному истончению БМК. В интерстиции можно обнаружить пенистые клетки, нагруженные мукополисахаридами и нейтральными жирами. Эта неспецифическая находка — следствие протеинурии, которая при этом заболевании может быть необычайно выраженной по непонятной причине.
При прогрессировании заболевания развиваются ФСГС и тотальный гломерулосклероз, а также происходят другие изменения, характерные для прогрессирующей патологии почек (например, склероз сосудов, атрофия канальцев и фиброз стромы). У большинства пациентов с развернутой формой этого заболевания выявляют характерные ультраструктурные изменения. Наблюдается неравномерное очаговое утолщение базальной мембраны, чередующееся с очагами истончения, а также растрескивание и расслоение плотной пластинки, что придает клубочкам вид плетеной корзины. Подобные изменения могут быть и в базальной мембране канальцев.
При пограничных изменениях базальной мембраны (или их отсутствии) проводят иммуногистохимическое исследование: при сцепленном с Х-хромосомой синдроме Альпорта антитела к а3-, а4-, а5-цепям коллагена типа IV не окрашиваются ни в гломерулярной, ни в канальцевой базальной мембране. Кроме того, результаты иммуногистохимического исследования (с антителами к а5-цепи) биоптатов кожи у этих пациентов отрицательные.
в) Клинические признаки. Наиболее типичным признаком является макро- или микрогематурия, часто сопровождающаяся скоплениями эритроцитов. Позже может развиться протеинурия, редко — нефротический синдром. Симптомы обычно появляются в возрасте от 5 до 20 лет, через 20-50 лет у мужчин развивается выраженная почечная недостаточность. С помощью высокочувствительных методов исследования слуха можно обнаружить незначительные нарушения.
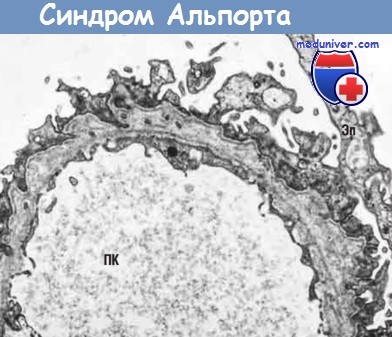
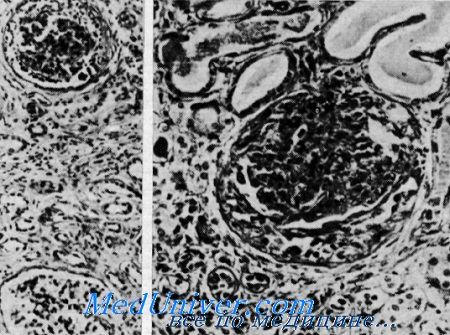
Сверху клубочек с утолщенной основной мембраной и слипшейся капсулой в противоположность почти нормальному клубочку внизу.
Видны редкие клубочки, имеющие полулупиую форму, расширенные канальцы, содержащие белковые цилиндры.
Утолщенная основная мембрана, окружающая атрофичные канальцы в области фиброза (из H. I. Krickstein et al.).
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) болезни тонких базальных мембран"
Оглавление темы "Патогенез заболеваний почек":- Механизм развития (патогенез) фокального сегментарного гломерулосклероза
- Механизм развития (патогенез) ВИЧ-ассоциированной нефропатии
- Механизм развития (патогенез) мембранопролиферативного гломерулонефрита (МПГН)
- Механизм развития (патогенез) нефропатии IgA - болезни Берже
- Механизм развития (патогенез) синдрома Альпорта
- Механизм развития (патогенез) болезни тонких базальных мембран
- Механизм развития (патогенез) хронического гломерулонефрита
- Механизм развития (патогенез) поражения почек при пурпуре Шенлейна-Геноха
- Механизм развития (патогенез) поражения почек при эндокардите
- Механизм развития (патогенез) поражения почек при сахарном диабете