
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Причины синдромов Прадера-Вилли и Ангельмана
Синдром Прадера-Вилли характеризуется умственной отсталостью, низким ростом, гипотонией, ожирением, маленькими размерами кистей и стоп и гипогонадизмом. В 60-70% случаев выявляется интерстициальная делеция сегмента q12 длинного плеча 15-й хромосомы fdel(15)(q11;q13)].
В большинстве случаев места разрывов совпадают, при этом длина делеции составляет 5 мегабаз. Поразительным является тот факт, что во всех случаях делеция происходит на отцовской 15-й хромосоме.
Пациенты с фенотипически отличающимся синдромом Ангельмана рождаются с делецией в этой же области 15-й хромосомы, но полученной от матери. Пациенты с синдромом Ангельмана также страдают умственной отсталостью, однако помимо этого наблюдаются атаксия, припадки и пароксизмальный смех. Из-за такого смеха и атаксии это заболевание образно назвали синдромом счастливой куклы. Сравнение этих синдромов четко демонстрирует «родительскую природу» эффектов функционирования генов.
Молекулярные основы этих двух синдромов можно понять в контексте импринтинга. Считается, что набор генов на материнской хромосоме 15q12 подвергается импринтингу (следовательно, инактивируется). Таким образом, функционирующие аллели предоставляются только отцовской хромосомой. Когда они утрачиваются на отцовской хромосоме в результате делеции, у пациента развивается синдром Прадера-Вилли.
И наоборот, определенный ген, который располагается в этой же области 15-й хромосомы, подвергается импринтингу на отцовской хромосоме. В норме активен только материнский аллель этого гена. Делеция этого гена на материнской 15-й хромосоме приводит к развитию синдрома Ангельмана. Молекулярные исследования цитогенетически нормальных пациентов с синдромом Прадера-Вилли выявили, что в некоторых случаях обе структурно нормальные 15-е хромосомы имеют материнскую природу. Наследование обеих хромосом в паре от одного из родителей называют унипаренталъной дисомией.
Конечный результат остается таким же: у пациента отсутствует функционирующий набор генов (не подвергшийся импринтингу) с 15-й хромосомы, полученной от отца. Можно предположить, что синдром Ангельмана также может возникать в результате унипарентальной дисо-мии отцовской 15-й хромосомы.
Молекулярные основы этих двух заболеваний, вызванных нарушением геномного импринтинга, сейчас тщательно изучают. Установлено, что при синдроме Ангельмана поражается ген UBE3A, кодирующий убиквитин-лигазу, которая участвует в катализе переноса активированного убиквитина, нацеленного на белковые субстраты.
Этот ген, расположенный в области 15q12, подвергается импринтингу на отцовской хромосоме и экспрессируется с материнского аллеля в специфических областях нормального головного мозга. Импринтинг в данном случае является тканеспецифическим, т.е. в большинстве тканей ген UBE3A экспрессируется с обоих аллелей.
В 10% случаев синдром Ангельмана развивается не вследствие импринтинга, а в результате точечной мутации материнского аллеля. Этот факт подчеркивает устойчивую связь синдрома Ангельмана с геном UBE3A. В отличие от синдрома Ангельмана, связь синдрома Прадера-Вилли с каким-либо одним геном не установлена. Напротив, считается, что в процесс вовлечены несколько генов, располагающихся между 15q11.2 и q13 (которые подвергаются импринтингу на материнской хромосоме и экспрессируются с отцовской).
К этим генам относится ген, кодирующий малый ядерный рибопротеин N, который контролирует генный сплайсинг и особенно широко экспрессируется в головном мозге и сердце. Считается, что утрата функции этого рибопротеина вносит свой вклад в развитие синдрома Прадера-Вилли. Молекулярная диагностика этих заболеваний основана на оценке состояния метилирования маркерных генов и методе FISH.
Значение импринтинга не ограничено редкими хромосомными заболеваниями. Эффекты, зависящие от «родительской природы», играют роль в развитии многих других наследственных заболеваний, в т.ч. болезни Хантингтона и миотонической дистрофии, а также в онкогенезе.
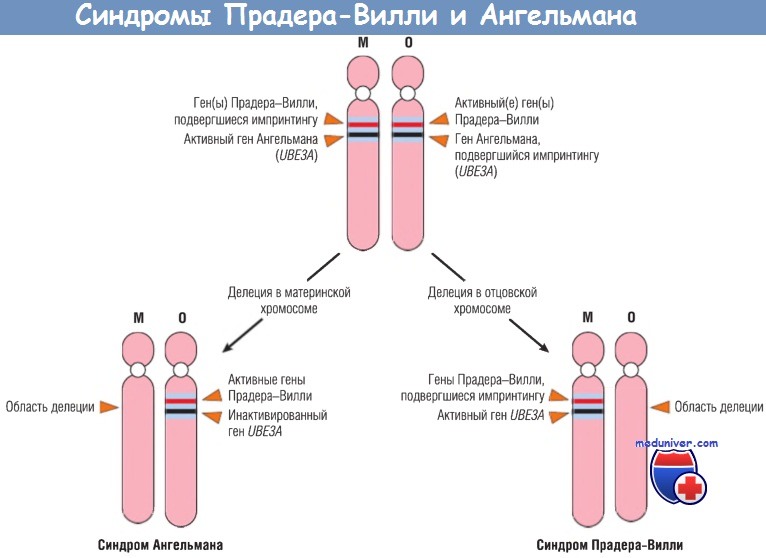
М — материнская хромосома; О — отцовская хромосома.
- Рекомендуем ознакомиться со следующей статьей "Болезни вызванные гонадным мозаицизмом"
Оглавление темы "Генетические болезни":- Причины, механизмы развития гермафродитизма и псевдогермафродитизма
- Болезни с мутацией типа тринуклеотидных повторов
- Причины синдрома ломкой Х-хромосомы и механизмы ее развития
- Болезни вызванные мутацией в митохондриальных генах
- Болезни вызванные геномным импритингом
- Причины синдромов Прадера-Вилли и Ангельмана
- Болезни вызванные гонадным мозаицизмом
- Молекулярные методы диагностики генетических болезней
- Показания для генетических исследований зародышевых клеток
- Показания для анализа генетических изменений клеток