
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Причины синдрома ломкой Х-хромосомы и механизмы ее развития
Синдром ломкой Х-хромосомы — прототип заболеваний, при которых мутации имеют вид длинных последовательностей повторяющихся тринуклеотидов. Хотя специфические нуклеотидные последовательности, которые подвергаются амплификации, при различных заболеваниях, включенных в данную группу (около 20), отличаются, в большинстве случаев вовлеченные в процесс последовательности содержат нуклеотиды G и С. Рассмотрим клинические признаки и механизм наследования синдрома ломкой Х-хромосомы, а также молекулярные нарушения, приводящие к данному заболеванию. Остальные заболевания этой группы будут описаны далее в этой и в других главах.
Данный синдром является второй по распространенности (после синдрома Дауна) генетической причиной умственной отсталости: его частота достигает 1 : 1550 среди мужчин и 1 : 8000 среди женщин. Этот синдром является сцепленным с Х-хромосомой заболеванием и характеризуется индуцибельной цитогенетической аномалией Х-хромосомы и необычной мутацией в гене FMR1. Цитогенетические изменения отличаются неравномерностью окрашивания или появлением как бы разрыва на длинном плече Х-хромосомы после культивирования клеток в среде без фолата.
Создается впечатление, что Х-хромосома «сломана», поэтому эту область назвали ломкой областью. Необходимо отметить, что в геноме человека обнаружено более 100 ломких областей. Многие из них, например область, обнаруживаемая при синдроме ломкой Х-хромосомы, чувствительны к недостатку фолата в среде, в то время как для других областей необходимы иные условия культивирования. Многие ломкие области обнаруживают у здоровых людей, поэтому значение таких областей все еще остается неизвестным.
При синдроме ломкой Х-хромосомы у больных мужчин в возрасте от 20 до 60 лет клинически проявляется умственная отсталость. Эти больные имеют типичный внешний вид: удлиненное лицо с большой нижней челюстью, большие оттопыренные ушные раковины и увеличенные яички (макроорхидизм). Переразгибание суставов, высокое небо и пролапс митрального клапана, наблюдаемые у некоторых больных, могут подтолкнуть врача к ошибочному диагностированию заболевания соединительной ткани. Перечисленные ранее симптомы присутствуют не всегда или едва заметны.
Единственный явный физический признак, который в 90% случаев присутствует у мужчин постпубертатного возраста с синдромом ломкой Х-хромосомы, — это макроорхидизм.
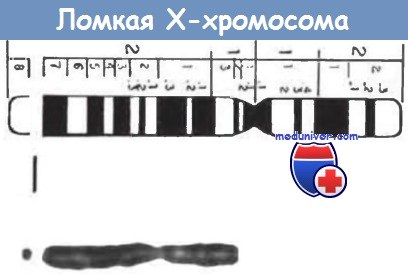
Видимость разрыва на длинном плече хромосомы при окрашивании.
Как все сцепленные с Х-хромосомой заболевания, синдром ломкой Х-хромосомы выявляют у мужчин. Однако анализ нескольких родословных позволил обнаружить некоторые закономерности наследования, нехарактерные для других сцепленных с Х-хромосомой рецессивных заболеваний:
- носители-мужчины. Приблизительно 20% мужчин, которые по результатам анализа родословной и молекулярных тестов являются носителями мутации ломкой Х-хромосомы, не имеют клинических и цитогенетических признаков заболевания. Такие мужчины могут передавать это заболевание своим внукам мужского пола через фенотипически нормальных дочерей;
- женщины с заболеванием. 30-50% женщин-носителей страдают этим заболеванием (имеют умственную отсталость). Это количество значительно больше, чем обычно отмечается при других сцепленных с Х-хромосомой заболеваниях;
- риск фенотипических проявлений. Риск зависит от положения носителя в родословной. Например, для братьев мужчины-носителя риск умственной отсталости составляет 9%, в то время как для его внуков — 40%;
- антиципация (проявление болезни в более молодом возрасте у представителей следующих поколений). Клинические признаки синдрома ломкой Х-хромосомы утяжеляются в каждом последующем поколении, т.к. мутация становится все более разрушительной при передаче ее от отца внукам и правнукам.
Эта необычная картина в течение многих лет озадачивала генетиков. Сейчас проводят молекулярные исследования, цель которых — изучение механизмов этого заболевания. Первым прорывом стало обнаружение с помощью линкерного анализа мутации на Xq27.3 в цитогенетически аномальной области. В этой области располагается ген FMR1, характеризующийся множественными тандемными повторами нуклеотидной последовательности CGG в 5'-нетранслируемой области. В нормальной популяции количество повторов CGG в гене FMR1 обычно составляет 6-53 (в среднем 29). Наличие клинической симптоматики и выявляемой цитогенетическими методами ломкой области ассоциируется с амплификацией повторов CGG. Таким образом, здоровые мужчины-носители и женщины-носители имеют от 60 до 200 повторов CGG. Экспансии такого размера называют премутациями.
У больных этим синдромом такие экспансии чрезвычайно длинные — от 200 до 4000 повторов (полные мутации). Считается, что полные мутации возникают при дальнейшей амплификации премутаций. Этот процесс представляет особый интерес. Мужчины-носители передают повторы потомству с небольшим изменением количества. Однако при передаче премутации от женщины-носителя существует очень большая вероятность значительной амплификации повторов, что приводит к умственной отсталости практически всех потомков мужского пола и 50% потомков женского пола. Таким образом, экспансия премутации до полной мутации происходит в процессе оогенеза, а не сперматогенеза. Это объясняет необычный характер наследования. Вероятность умственной отсталости гораздо выше у внуков мужчины-носителя, чем у его братьев, т.к. у внуков существует большой риск наследования от своего деда премутации, ампли-фицированной до полной мутации в яйцеклетке матери.
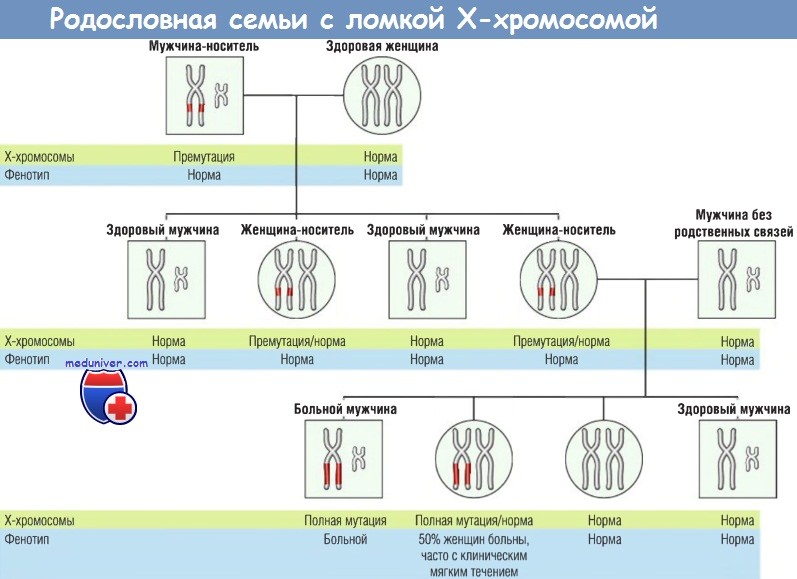
Обратите внимание, что в первом поколении все сыновья здоровы, а дочери являются носителями.
Во время оогенеза у женщины-носителя премутация превращается в мутацию.
Таким образом, в следующем поколении все дети мужского пола, которые наследуют Х-хромосому с полной мутацией, являются больными.
Однако только 50% детей женского пола, унаследовавших полную мутацию, являются больными, и часто болезнь протекает мягко.
Для братьев мужчины-носителя, находящихся «выше» в родословной, риск наследования полной мутации гораздо меньше. Эти детали наследования также объясняют явление антиципации — феномена, замеченного клиническими генетиками, в который молекулярные генетики, однако, не верили до тех пор, пока не были обнаружены мутации типа тринуклеотидных повторов. Открытым остается вопрос о том, почему заболевание развивается только у 50% женщин с полной мутацией. Возможно, это связано с неблагоприятной лайонизацией у женщин с клиническими признаками заболевания (т.е. большое количество клеток содержит активную Х-хромосому с мутацией). Последние исследования указывают на то, что премутации не так уж безобидны, как предполагалось ранее.
У 30% женщин, являющихся носителями премутаций, в возрасте до 40 лет обнаруживается преждевременная недостаточность яичников, а у 30% мужчин, являющихся носителями премутаций, начиная с 50 лет развивается прогрессирующий нейродегенеративный синдром. Этот синдром, который называют тремор/атаксия, ассоциированные с ломкой Х-хромосомой, характеризуется наличием интенционного тремора и мозжечковой атаксии и может перерасти в паркинсонизм. Однако симптомы при носительстве премутаций развиваются позднее и являются более мягкими.
Молекулярные механизмы развития умственной отсталости и других соматических нарушений связаны с утратой функции FMRP. Как уже упоминалось ранее, нормальный ген FMR1 содержит до 53 повторов CGG в своей 5'-нетранслируемой области. Если количество тринуклеотидных повторов превышает 230, ДНК всей 5'-нетранслируемой области подвергается аномальному метилированию. Метилирование достигает промоторной области гена, приводя к транскрипционной супрессии FMR1. Считается, что фенотипические изменения связаны с отсутствием FMRP.
FMRP широко экспрессируется в цитоплазме всех здоровых тканей, однако больше всего его обнаруживают в головном мозге и яичках — двух органах, страдающих при данном заболевании в наибольшей мере. Только сейчас становится понятной функция FMRP в головном мозге. FMRP является связывающим РНК белком, ассоциированным с полисомами. В отличие от других клеток, синтез белка в нейронах происходит как в перинуклеарной цитоплазме, так и в дендритах. Последние данные указывают на то, что FMRP поступает из цитоплазмы в ядро, где соединяется со специфическими транскриптами мРНК.
Оттуда комплекс FMRP-мPHK транспортируется в дендриты нейронов ближе к синапсу. В дендриты транспортируются не все виды мРНК, а только те, которые кодируют белки, участвующие в регуляции синаптической функции. В области синапса FMRP подавляет синтез белка со связанной с ним мРНК посредством сигнального пути, использующего метаботрофичные рецепторы к глутамату группы 1. При синдроме ломкой Х-хромосомы уменьшение количества FMRP приводит к увеличению трансляции связанных с ним мРНК в области синапса. Такой дисбаланс, в свою очередь, обусловливает постоянные изменения синаптической активности и появление основного клинического симптома — умственной отсталости.
Идентифицировать это заболевание позволило выявление аномального кариотипа, но методом диагностики остается обнаружение повторов с помощью ПЦР Для дифференцировки премутаций и мутаций в ДНК в пренатальном и постнатальном периодах используют саузерн-блоттинг. Таким образом, этот метод ценен не только для диагностики, но и для проведения генетического консультирования.
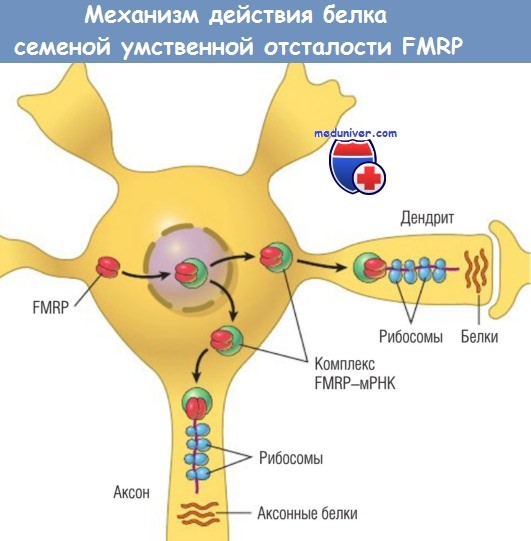
мРНК — матричная рибонуклеиновая кислота
Видео этиология, патогенез синдрома ломкой Х хромосомы (синдрома Мартина-Белл)
- Рекомендуем ознакомиться со следующей статьей "Болезни вызванные мутацией в митохондриальных генах"
Оглавление темы "Генетические болезни":- Причины, механизмы развития гермафродитизма и псевдогермафродитизма
- Болезни с мутацией типа тринуклеотидных повторов
- Причины синдрома ломкой Х-хромосомы и механизмы ее развития
- Болезни вызванные мутацией в митохондриальных генах
- Болезни вызванные геномным импритингом
- Причины синдромов Прадера-Вилли и Ангельмана
- Болезни вызванные гонадным мозаицизмом
- Молекулярные методы диагностики генетических болезней
- Показания для генетических исследований зародышевых клеток
- Показания для анализа генетических изменений клеток
- 5 причин, из-за которых вы не худеете