
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития синдрома гипер-IgM
При синдроме гипер-IgM иммунная система пациентов продуцирует IgM-антитела, однако способность продуцировать антитела других классов — IgG, IgA и IgE — нарушена. В настоящее время известно, что при данном заболевании страдает способность хелперных Т-клеток генерировать активирующие сигналы для В-клеток и макрофагов.
Как было указано ранее в этой главе, для многих функций хелперных Т-клеток CD4+ необходимо взаимодействие молекулы CD40L (называемой также CD154), экспрессированной активированными антигеном Т-клетками, с молекулой CD40, которую экспрессируют В-клетки, макрофаги и дендритные клетки.
Это взаимодействие инициирует переключение классов Ig и созревание аффинности в В-клетках и стимулирует антимикробные функции макрофагов. Приблизительно 70% индивидов с синдромом гипер-IgM имеют сцепленное с Х-хромосомой заболевание, вызванное мутацией гена, кодирующего CD40L и расположенного на хромосоме Xq26. У оставшейся части пациентов болезнь наследуется по аутосомно-рецессивному типу.
Большинство этих пациентов имеют мутации гена, кодирующего CD40, или фермент, называемый индуцированной активацией дезаминазой, ДНК-редактирующей цитозиндезаминазой, необходимой для переключения класса и созревания аффинности.
Сыворотка лиц с данным синдромом содержит нормальное или повышенное количество IgM, однако в ней отсутствуют IgA или IgE и определяется крайне низкий уровень IgG. Число В- и Т-клеток близко к норме. Многие IgM-антитела реагируют с элементами крови, вызывая аутоиммунную гемолитическую анемию, тромбоцитопению и нейтропению.
У пожилых пациентов может наблюдаться неконтролируемая пролиферация плазматических клеток, продуцирующих IgM, с инфильтрацией ЖКТ. Хотя пролиферирующие В-клетки являются поликлональными, обширная инфильтрация может послужить причиной их смерти.
Клиническая картина у пациентов с синдромом гипер-IgM характеризуется рецидивирующими пиогенными инфекциями вследствие низкого уровня опсонизирующих IgG-антител. Кроме того, лица с мутациями CD40L восприимчивы к пневмонии, вызываемой внутриклеточным микроорганизмом P. jiroveci, в результате дефекта клеточного иммунитета.
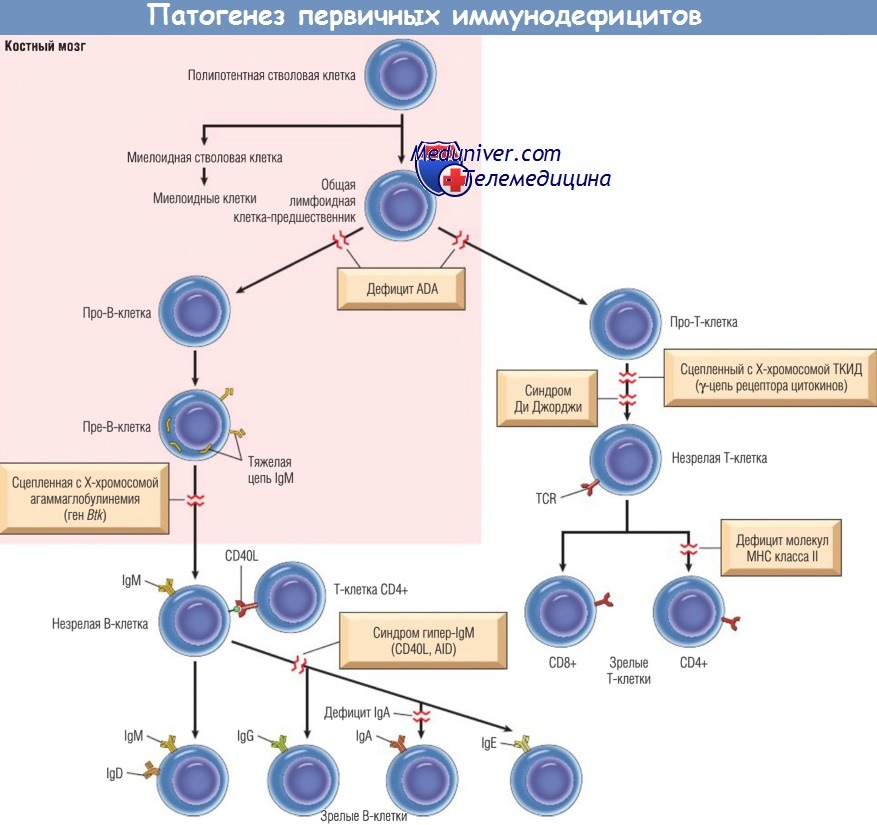
Для некоторых расстройств в скобках указан ответственный за них ген.
ADA — аденозиндезаминаза; AID — дезаминаза, индуцированная активацией; CD40L — лиганд CD40 (известный также как CD154);
Ig — иммуноглобулин; МНС — главный комплекс гистосовместимости; TCR — Т-клеточный рецептор; ТКИД — тяжелый комбинированный иммунодефицит.
- Рекомендуем ознакомиться со следующей статьей "Механизмы развития синдрома Ди Джорджи (гипоплазии тимуса)"
Оглавление темы "Заболевания иммунной системы":- Механизмы развития синдрома гипер-IgM
- Механизмы развития синдрома Ди Джорджи (гипоплазии тимуса)
- Механизмы развития тяжелого комбинированного иммунодефицита (ТКИД)
- Механизмы развития иммунодефицита с тромбоцитопенией и экземой (синдрома Вискотта-Олдрича)
- Механизмы развития генетической недостаточности системы комлемента
- Вторичные иммунодефициты и их характеристика
- Распространенность СПИД (ВИЧ) и группы риска
- Свойства ВИЧ и его строение
- Механизмы развития ВИЧ-инфекции - жизненный цикл ВИЧ
- Механизмы иммунодефицита при ВИЧ-инфекции