
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Морфология и механизмы развития опухоли Вильмса
Опухоль Вильмса возникает у 1 из 10 тыс. детей в США и является наиболее часто встречающейся первичной опухолью почек у детей и четвертой по частоте злокачественной опухолью в популяции детей в США. Пик возникновения опухоли приходится на промежуток между 2 и 5 годами, а 95% опухолей появляются до достижения ребенком 10-летнего возраста.
Приблизительно в 5-10% случаев опухоль Вильмса поражает обе почки или одновременно (синхронные опухоли), или последовательно (метахронные опухоли). Двухсторонняя опухоль Вильмса возникает в среднем на 10 мес раньше, чем опухоль, поражающая одну почку, и, как предполагается, эти пациенты имеют герминативную мутацию одного из генов, ответственных за развитие опухоли Вильмса.
Биология этой опухоли отражает несколько важных характеристик, присущих детским опухолям: взаимосвязь врожденных пороков развития с высоким риском неоплазий, гистологическое сходство опухоли и развивающегося органа, значительную эффективность лечения, а также роль предраковых поражений и справедливость гипотезы «два удара» с участием рецессивных генов-супрессоров опухолей. Рост показателей эффективности терапии опухоли Вильмса (с 30% несколько десятилетий назад до 85% в настоящее время) является одним из значительных успехов современной педиатрической онкологии.
Патогенез и генетические аспекты. Риск развития опухоли Вильмса повышен в некоторых группах врожденных пороков развития (например, при WAGR-синдроме, синдроме Дениса-Драша, синдроме Беквита-Видемана) и связан с определенными хромосомными локусами. Хотя доля опухолей, развивающихся в рамках данных синдромов, составляет не более 10%, их изучение обеспечило более глубокое понимание биологии опухоли Вильмса.
Первая группа пациентов имеет WAGR-синдром, характеризующийся аниридией, пороками развития гениталий и умственной отсталостью. При этом вероятность развития опухоли Вильмса составляет 33%. Дети с WAGR-синдромом несут конституциональную (на уровне зародышевых клеток) делецию хромосомы 11р13. При исследовании были обнаружены ген WT1, ассоциированный с опухолью Вильмса, и близко расположенный доминантный ген аниридии РАХ6, локализующиеся на хромосоме 11р13. У пациентов с делецией, затрагивающей РАХ6, и нормальной функцией WT1 развивается спорадическая аниридия, но не увеличивается риск развития опухоли Вильмса.
Конституциональная делеция гена WT1 при WAGR-синдроме является первым ударом; развитие опухоли Вильмса у этих пациентов часто коррелирует с нонсенс-мутацией или мутацией со сдвигом рамки считывания во втором аллеле WT1 (второй удар).
Вторая группа пациентов имеет синдром Дениса-Драша, который характеризуется дисгенезией гонад (мужской псевдогермафродитизм) и ранней нефропатией, ведущей к почечной недостаточности. Эта группа отличается высоким риском развития опухоли Вильмса (« 90%). Клубочковую патологию при этом синдроме описывают как диффузный мезангиальный склероз (см. главу 20). Как и при WAGR-синдроме, у этих пациентов отмечается патология WT1, которая проявляется доминантно-негативной миссенс-мутацией в цинксвязывающей области гена WT1, что оказывает влияние на его ДНК-связывающие свойства. Эта мутация изменяет функцию оставшегося аллеля дикого типа, что проявляется только мочеполовыми аномалиями, но не опухолевым ростом. Опухоли Вильмса на фоне синдрома Дениса-Драша развиваются только при биаллельной инактивации WT1.
Помимо опухоли Вильмса эти пациенты также имеют риск развития опухолей из зародышевых клеток — гонадобластом, вероятно вследствие нарушения нормального формирования гонад.

WT1 кодирует ДНК-связывающий фактор транскрипции, который экспрессируется во время эмбриогенеза в пределах некоторых тканей, включая почки и половые железы. Белок WT1 важен для нормального формирования почек и половых желез. WT1 имеет множество партнеров для связывания, и их выбор определяет, будет WT1 функционировать как активатор или репрессор транскрипции. Были выявлены многочисленные транскрипционные мишени WT1, включающие гломерулярные подоцит-специфические белки и гены, ассоциированные с индукцией дифференцировки клеток. Несмотря на важность WT1 в процессах нефрогенеза и его определенную роль как гена-супрессора опухолей, только 10% пациентов со спорадическими (несиндромными) опухолями Вильмса имеют мутации WT1. Это позволяет предположить, что большинство таких опухолей возникает генетически различными путями.
Дети с синдромом Беквита-Видемана также имеют высокий риск развития опухоли Вильмса, но клинически отличаются от предыдущих двух групп пациентов и характеризуются увеличением отдельных органов (органомегалией), макроглоссией, гемигипертрофией, пупочной грыжей и патологическим увеличением клеток коры надпочечников (надпочечниковой цитомега-лией). Синдром Беквита-Видемана послужил моделью для изучения неклассического механизма развития опухолей у человека — геномного импринтинга. Генетический локус, вовлеченный у таких больных в патологический процесс, располагается на 11-й хромосоме в сегменте р15.5 (WT2), дистальнее локуса WT1.
Эта область содержит несколько генов, которые в норме экспрессируются только с одним из двух родительских аллелей, при этом второй родительский гомолог транскрипционно не активирован за счет метилирования промоторной области.
В отличие от WAGR-синдрома и синдрома Дениса-Драша генетическая основа синдрома Беквита-Видемана значительно более гетерогенна, т.к. во всех случаях вовлечен не только ген 11р15.5. Более того, фенотип синдрома Беквита-Видемана, включающий предрасположенность к возникновению опухолей, зависит от специфических нарушений области импринтинга WT2. Один из генов этой области — IGF2, который в норме экспрессируется только отцовским аллелем, в то время как материнский аллель подвергается импринтингу.
В некоторых опухолях Вильмса можно обнаружить утрату импринтинга (т.е. экспрессию IGF2 с материнским аллелем), что ведет к чрезмерному образованию белка IGF-2. В других случаях происходит селективная делеция импринтинга материнского аллеля, комбинированная с дупликацией транскрипционно активного отцовского аллеля (унипарентальной дисомией) в опухоли, которая имеет идентичный эффект с точки зрения гиперэкспрессии IGF-2. Поскольку IGF-2 является эмбриональным фактором роста, этим можно объяснить наличие у пациентов с синдромом Беквита-Видемана клинического симптома избыточного роста, а также повышенного риска появления опухоли Вильмса.
Из всех генов области импринтинга WT2 самую сильную связь с предрасположенностью к развитию опухолей при синдроме Беквита-Видемана имеет ген IGF2. В субпопуляции людей с этим синдромом также отмечается мутации регулятора клеточного цикла CDKN1С (также известного как р57 или KIP2), однако у этих пациентов риск развития опухоли Вильмса значительно ниже. Пациенты с синдромом Беквита-Видемана также имеют повышенный риск развития гепатобластомы, панкреатобластомы, опухоли коры надпочечников и рабдомиосаркомы.
Недавние генетические исследования определили роль b-катенина в развитии опухоли Вильмса. Следует напомнить, что b-катенин принадлежит к сигнальному пути WNT. Доминантно-негативные мутации гена, кодирующего b-катенин, были обнаружены в 10% спорадических опухолей Вильмса. Существует значительное перекрытие мутаций WT1 и b-катенина, что предположительно определяет синергизм этих событий в генезе опухоли Вильмса. Предполагают, что нефрогенные островки — это клетки-предшественники опухоли Вильмса, которые иногда обнаруживают в почечной паренхиме, прилежащей к опухоли. Нефрогенные островки появляются в 25-40% случаев при односторонней опухоли и в 100% случаев при двухсторонней опухоли Вильмса. Во многих случаях нефрогенные островки имеют одинаковые генные мутации с прилежащей опухолью Вильмса, что подтверждает их пренеопластический статус.
Внешний вид нефрогенных островков варьирует от объемных масс (гиперпластических островков) до склерозированных островков, состоящих преимущественно из фиброзной ткани, в которой периодически встречаются незрелые канальцы или клубочки. Необходимо отмечать наличие в препарате нефрогенных островков, т.к. это повышает риск развития опухоли Вильмса в контралатеральной почке и определяет частоту и регулярность наблюдений на долгие годы.
Морфология. Макроскопически опухоль Вильмса чаще всего имеет вид большого, единичного, хорошо отграниченного новообразования, хотя в 10% случаев на момент постановки диагноза опухоль может быть двухсторонней или мультицентричной. На разрезе опухоль имеет серый оттенок, обладает мягкой консистенцией, возможны очаги кровоизлияний, кистозной дегенерации и некроза.
Микроскопически опухоль Вильмса характеризуется наличием структур, напоминающих различные стадии нефрогенеза. В большинстве случаев можно обнаружить классическую трехфазную комбинацию зародышевых, стромальных и эпителиальных клеток, хотя количественное соотношение этих клеток в опухоли может быть различным. Пласты мелких клеток с некоторыми отличительными особенностями составляют бластный компонент. Эпителиальная дифференцировка обычно принимает вид рудиментарных канальцев или клубочков. Клетки стромы часто имеют фибробластическую или миксоидную природу, однако нередко наблюдается дифференцировка в клетки поперечнополосатых мышц.
Иногда можно выявить и другие гетерологичные элементы, включая плоский или железистый эпителий, клетки гладких мышц, жировую, хрящевую, костную или нейрогенную ткань.
Приблизительно 5% опухолей содержат очаги анаплазии (клетки с крупным гиперхромным плеоморфным ядром и патологическими фигурами митоза). Анаплазия связана с мутациями р53 и развитием резистентности к химиотерапии. Вспомним, что р53 запускает процессы апоптоза в клетке в ответ на повреждение ДНК. Утрата функции р53 объясняет устойчивость анапластических клеток к цитотоксической химиотерапии.
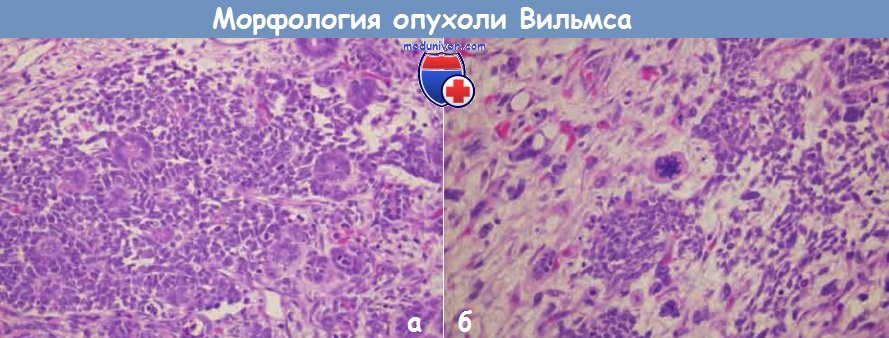
и рудиментарные канальцы, представляющие собой эпителиальный компонент. Хотя видны множественные фигуры митоза, признаков атипии нет.
(б) Локальная анаплазия в других областях той же опухоли Вильмса характеризуется клетками с гиперхромными плеоморфными ядрами и фигурами митоза.
- Рекомендуем ознакомиться со следующей статьей "Клиника и прогноз опухоли Вильмса у ребенка"
Оглавление темы "Патогенез детских болезней":- Причины и факторы риска синдрома внезапной детской смерти (СВДС)
- Морфология и механизмы развития синдрома внезапной детской смерти (СВДС)
- Виды доброкачественных опухолей у детей
- Виды злокачественных опухолей у детей
- Причины, морфология и стадии нейробластомы
- Клиника и прогноз нейробластомы у ребенка
- Морфология и механизмы развития опухоли Вильмса
- Клиника и прогноз опухоли Вильмса у ребенка