
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) острого лимфоцитарного лейкоза
Опухолевые клетки при острой лимфобластной лейкемии/лимфоме (ОЛЛ) представляют собой незрелые В-клетки (пре-В-клетки) или Т-клетки (пре-Т-клетки), называемые лимфобластами. Около 85% всех ОЛЛ — это В-клеточная ОЛЛ (В-ОЛЛ), в типичных случаях проявляющаяся как детская острая лейкемия. Реже встречающаяся Т-клеточная ОЛЛ (Т-ОЛЛ) наблюдается преимущественно у подростков в виде тимусной лимфомы. Клиническая картина В-ОЛЛ и Т-ОЛЛ близка: например, В-ОЛЛ может принимать необычную форму опухолевой массы в коже или костях, а Т-ОЛЛ во многих случаях изначально или со временем дает клиническую картину лейкемии. Из-за морфологического и клинического сходства различные формы ОЛЛ рассмотрены вместе.
Острый лимфобластный лейкоз (ОЛЛ) — наиболее частая злокачественная опухоль у детей. Ежегодно в США диагностируют 2500 новых случаев, и большинство из них регистрируют у детей до 15 лет. Частота острого лимфобластного лейкоза (ОЛЛ) у лиц с белым цветом кожи в 3 раза выше, чем у лиц с темным цветом кожи. Заболевание чаще встречается у мальчиков. Из всех этнических групп наибольшая частота отмечена у испаноязычных американцев. Пик заболеваемости приходится на 3-летний возраст, возможно потому, что количество пре-В-клеток в нормальном костном мозге больше всего именно в этом возрасте. Пик заболеваемости Т-ОЛЛ наблюдается в подростковом возрасте, когда тимус достигает максимального размера. В-ОЛЛ и Т-ОЛЛ у взрослых регистрируют реже.
а) Морфология. При лейкемии костный мозг отличается гиперклеточностью и обилием лимфобластов, плотные скопления которых замещают нормальные элементы костного мозга. Медиастинальные опухолевые массы в тимусе встречаются в 50-70% случаев Т-ОЛЛ. В этой ситуации более вероятна связь с лимфаденопатией и спленомегалией. И при В-ОЛЛ, и при Т-ОЛЛ опухолевые клетки имеют скудную агранулярную базофильную цитоплазму и более крупные ядра, чем у малых лимфоцитов. Ядерный хроматин имеет тонкую точечную структуру, ядрышки либо отсутствуют, либо незаметны. Во многих случаях ядерная мембрана глубоко разделена, что придает ей инвагинированный вид. В соответствии с агрессивным клиническим течением в клетках высока частота митоза. Как и в случае других быстрорастущих опухолей, рассеянные в опухоли макрофаги, поглотившие апоптозные опухолевые клетки, могут создавать картину «звездного неба».
Вследствие неодинаковой реакции опухолей на химиотерапию ОЛЛ необходимо отличать от острой миелоидной лейкемии (ОМЛ) — неоплазии, состоящей из незрелых миелоидных клеток, но которая может иметь идентичные признаки и симптомы. По сравнению с миелобластами лимфобласты имеют более конденсированный ядерный хроматин, менее заметные ядрышки и меньшее количество цитоплазмы, в которой обычно отсутствуют гранулы. Однако эти морфологические различия неабсолютны и окончательный диагноз базируется на окрашивании с помощью антител, специфичных для антигенов В- и Т-клеток.
Гистохимический метод позволяет установить, что в отличие от миелобластов лимфобласты дают отрицательную реакцию на миелопероксидазу и часто содержат PAS-положительный цитоплазматический материал.
б) Иммунофенотип. Иммуноокрашивание для выявления терминальной дезоксинуклеотидилтрансферазы (специализированной ДНК-полимеразы, экспрессированной только пре-В- и пре-Т-лимфобластами) дает положительный результат в 95% случаев. В-ОЛЛ и Т-ОЛЛ различают по окрашиванию специфических В- и Т-клеточных маркеров (см. далее).
В-ОЛЛ останавливается на разных стадиях развития пре-В-клеток. Лимфобласты обычно экспрессируют общий В-клеточный маркер CD19 и фактор транскрипции РАХ5, а также CD10. При очень незрелых В-клетках CD10 отсутствует. С другой стороны, более зрелые пре-В-клетки ОЛЛ экспрессируют CD10, CD19, CD20 и цитоплазматическую тяжелую цепь IgM (u-цепь).
Т-ОЛЛ также останавливается на разных стадиях развития Т-клеток. В большинстве случаев эти клетки дают положительную реакцию на CD1, CD2, CD5 и CD7. Незрелые пре-Т-клеточные опухоли обычно лишены поверхностных CD3, CD4 и CD8, тогда как более зрелые Т-клетки эти маркеры экспрессируют.
в) Молекулярный патогенез. Около 90% ОЛЛ характеризуются количественными или структурными изменениями хромосом. Наиболее часто встречается гиперплоидия (> 50 хромосом), однако возможны также гипоплоидия и сбалансированные хромосомные транслокации. Эти изменения нередко коррелируют с иммунофенотипом и иногда имеют прогностическое значение. Так, гипер- и гиподиплоидия присутствуют только при В-ОЛЛ. Кроме того, В-ОЛЛ и Т-ОЛЛ ассоциируются с разными наборами транслокаций, что указывает на патогенетические различия. Профиль РНК, определяемый с помощью генных чипов, свидетельствует о корреляции определенных хромосомных транслокаций с уникальными формами экспрессии генов.
Многие из хромосомных аберраций, обнаруживаемых при ОЛЛ, нарушают регуляцию экспрессии и функции факторов транскрипции, необходимых для нормального развития В- и Т-клеток. До 70% Т-ОЛЛ характеризуются мутациями с усилением функции NOTCH1, гена, важного для развития Т-клеток. С другой стороны, при В-ОЛЛ нередко обнаруживаются мутации с утратой функции генов, необходимых для развития В-клеток, например РЛХ5, Е2А и EBF, или сбалансированная транслокация t(12;21) генов TEL и AML1, необходимых для очень ранних гемопоэтических клеток-предшественников. Все эти мутации, видимо, нарушают дифференцировку лимфоидных клеток-предшественников и способствуют остановке созревания. Как будет описано далее, подобное происходит и при генезе ОМЛ.
В соответствии с теорией многоступенчатого канцерогенеза для развития ОЛЛ одиночных мутаций недостаточно. Этот вывод основан на результатах исследования идентичных близнецов с конкордантной В-ОЛЛ. В этих редких случаях ОЛЛ оба близнеца имеют общую хромосомную аберрацию и источником ОЛЛ служит одиночный клон, переносимый от одного близнеца другому in utero. Несмотря на присутствие лейкомогенной аберрации при рождении, клинически ОЛЛ у таких пациентов чаще всего проявляется в возрасте 4-12 лет. Такой удлиненный продромальный период обусловлен, скорее всего, существованием «предлейкемического» клона, в котором перед развитием ОЛЛ должны произойти дополнительные мутации. Идентичность этих комплементарных мутаций неполная, однако обычно присутствуют аберрации, повышающие пролиферацию и выживаемость лимфоцитов (например, активирующие мутации тирозинкиназ).
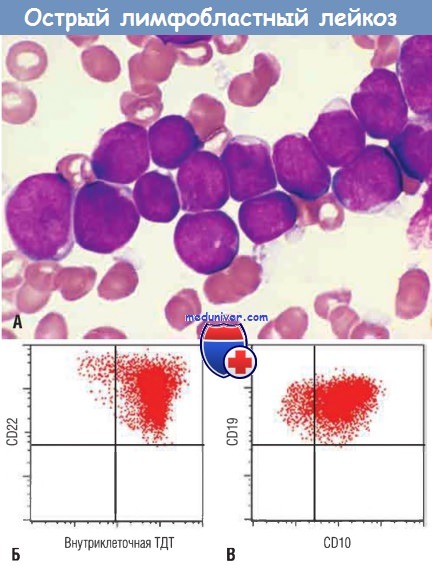
Лимфобласты с конденсированным ядерным хроматином, небольшими ядрышками и скудной агранулярной базофильной цитоплазмой.
(Б) Фенотип ОЛЛ определен с помощью проточной цитометрии.
Обратите внимание, что лимфобласты (красные точки) экспрессируют терминальную дезоксинуклеотидилтрансферазу (ТДТ) и В-клеточный маркер CD22.
(В) Те же клетки несут два других маркера — CD10 и CD19, обычно экспрессируемые пре-В-лимфобластами. Следовательно, это В-ОЛЛ.
г) Клинические признаки. Хотя ОЛЛ и ОМЛ генетически и иммунофенотипически различаются, клинически они очень сходны. В обоих случаях накопление неопластических бластов в костном мозге подавляет нормальный гемопоэз в результате физического сдавления, конкуренции за факторы роста и других малоизученных механизмов.
Общие проявления и признаки острого лимфобластного лейкоза (ОЛЛ) и острого миелобластного лейкоза (ОМЛ):
- резкое, бурное начало с появлением первых симптомов в течение нескольких дней или недель;
- симптомы, связанные с угнетением функции костного мозга, включая утомляемость, обусловленную анемией; лихорадка, вызванная инфекцией вследствие нейтропении; кровоточивость вследствие тромбоцитопении;
- проявления, обусловленные ростом опухоли и неопластической инфильтрацией (чаще наблюдаются при ОЛЛ), включая боль в костях вследствие экспансии костного мозга и инфильтрации поднадкостничного пространства; генерализованная лимфаденопатия, спленомегалия и гепатомегалия; увеличение тестикул; осложнения, связанные со сдавливанием крупных сосудов и дыхательных путей в средостении (при Т-ОЛЛ);
- проявления со стороны центральной нервной системы, например головная боль, рвота и парезы, возникающие в результате распространения процесса на оболочки головного мозга (эти проявления более характерны для ОЛЛ).
д) Прогноз. Терапия ОЛЛ у детей наиболее успешна. При «агрессивной» химиотерапии полная ремиссия отмечается в 95% случаев, а излечение — в 75-85%. Однако, несмотря на эти достижения, ОЛЛ остается основной причиной смерти от злокачественных опухолей среди детей. ОЛЛ у взрослых удается вылечить лишь в 35-40% случаев.
С плохим прогнозом ассоциируются следующие факторы:
(1) возраст до 2 лет (в значительной степени из-за связи ОЛЛ у детей с транслокацией гена MLL);
(2) манифестация в подростковом или взрослом возрасте;
(3) количество бластов в периферической крови свыше 100 тыс., что, вероятно, отражает опухолевую активность;
(4) наличие особых хромосомных аберраций, в частности транслокации t(9;22) (филадельфийской хромосомы), которая присутствует лишь у 3% детей с ОЛЛ, однако у взрослых частота достигает 25%. О плохом прогнозе свидетельствует наличие после лечения В-ОЛЛ и Т-ОЛЛ остаточного заболевания на молекулярном уровне, что стимулирует проведение новых клинических исследований.
Благоприятные прогностические факторы:
(1) возраст от 2 до 10 лет;
(2) низкое количество лейкоцитов;
(3) гиперплоидия;
(4) трисомия по 4, 7 и 10-й хромосомам;
(5) присутствие транслокации t(12;21).
Большинство хромосомных аберраций при остром лимфобластном лейкозе (ОЛЛ) изменяет функцию факторов транскрипции, но транслокация t(9;22) создает гибридный ген, кодирующий конститутивно активную тирозинкиназу BCR-ABL. При В-ОЛЛ тирозинкиназа BCR-ABL с молекулярной массой 190 кДа обладает более высокой активностью, чем тирозинкиназа BCR-ABL с молекулярной массой 210 кДа, обнаруживаемая при хронической миелоидной лейкемии. Лечение ОЛЛ с транслокацией t(9;22) ингибиторами тирозинкиназы BCR-ABL эффективно, однако быстро следует рецидив, поскольку приобретенные мутации BCR-ABL делают опухолевые клетки лекарственно резистентными. При В-ОЛЛ мутации BCR-ABL происходят часто. Этот феномен приписывают геномной нестабильности, способствующей клиническому прогрессированию болезни и резистентности к терапии у многих агрессивных злокачественных опухолей.
- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) хронического лимфоцитарного лейкоза"
Оглавление темы "Патогенез болезней":- Механизмы развития (патогенез) метастазов опухолей в сердце
- Механизмы развития (патогенез) осложнений трансплантации сердца
- Образование клеток крови (дифференцировка)
- Механизмы развития (патогенез) лейкопении и агранулоцитоза
- Механизмы развития (патогенез) лейкоцитоза
- Механизмы развития (патогенез) лимфаденита
- Механизмы развития (патогенез) неоплазий лейкоцитов
- Механизмы развития (патогенез) лимфоцитарного лейкоза и лимфомы
- Механизмы развития (патогенез) острого лимфоцитарного лейкоза
- Механизмы развития (патогенез) хронического лимфоцитарного лейкоза