
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) митохондриальной энцефаломиопатии
Многие наследственные заболевания, связанные с нарушениями процесса окислительного фосфорилирования в митохондриях, чаще всего проявляются как заболевания мышц, второе место занимает поражение ЦНС. Рассмотренная ранее в этой главе атаксия Фридрейха, обусловленная мутациями гена, кодирующего фратаксин, тоже относится к митохондриальным болезням.
а) Синдром митохондриальной энцефаломиопатии, лактат-ацидоза и инсультоподобных эпизодов. Митохондриальная энцефаломиопатия, лактат-ацидоз и инсультоподобные эпизоды (MELAS) — наиболее частый неврологический синдром, вызванный патологией митохондрий. Заболевание характеризуется рецидивирующими эпизодами острого неврологического дефицита с когнитивной дисфункцией, мышечной слабостью и лактат-ацидозом. Во время инсультоподобных эпизодов наблюдается обычно обратимая симптоматика, не коррелирующая с поражением определенного артериального бассейна.
При патоморфологическом исследовании выявляются очаги инфарктов с пролиферацией сосудов и фокусами кальцификации. Наиболее частые мутации генов, приводящие к MELAS, затрагивают транспортную РНК; также описаны мутации кодирующих генов.
б) Миоклоническая эпилепсия с рваными красными мышечными волокнами. Миоклоническая эпилепсия с рваными красными мышечными волокнами (MERRF) — наследуемое по материнской линии заболевание, характеризующееся миоклониями, судорожными приступами и миопатией. Важным симптомом является атаксия, обусловленная гибелью нейронов в структурах системы мозжечка (включая комплекс ядер нижней оливы в продолговатом мозге, кору мозжечка и его глубинные ядра). Большинство случаев MERRF вызваны мутациями транспортной РНК, которые отличаются от мутаций, лежащих в основе MELAS. У некоторых пациентов эти заболевания могут сочетаться.
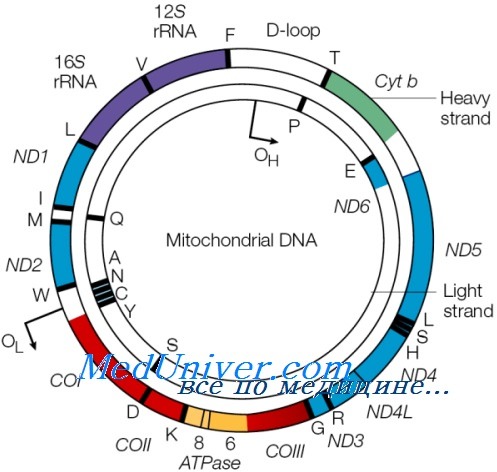
в) Синдром Лея. Синдром Лея (подострая некротизирующая энцефалопатия) — это заболевание раннего детского возраста, которое характеризуется лактат-ацидозом, задержкой психомоторного развития, проблемами с кормлением, судорожным синдромом, парезами глазодвигательных мышц и слабостью с гипотонусом. Летальный исход наступает обычно через 1-2 года.
При гистологическом исследовании выявляют мультифокальные, почти симметричные участки деструкции мозговой ткани, имеющие губчатый вид, и пролиферация сосудов. Наблюдается преимущественное поражение околоводопроводного серого вещества среднего мозга, покрышки моста и перивентрикулярных отделов таламуса и гипоталамуса.
Выявлено множество мутаций, каузальных для синдрома Лея, включая мутации ядерной и митохондриальной ДНК. Обнаружены различные мутации компонентов комплексов окислительного фосфорилирования, кодируемых митохондриальной ДНК, и мутации митохондриальной транспортной РНК, однако в этих случаях связь между генотипом и фенотипом, скорее всего, отсутствует. Интересно, что точечная мутация митохондриального гена, кодирующего АТФазную субъединицу комплекса V, вызывает развитие наследуемого по материнской линии синдрома Лея, при котором в клетках содержится большое количество мутантной митохондриальной ДНК.
Однако при наличии большего количества нормальных митохондрий синдром Лея с клинической и морфологической точек зрения развивается в другом ключе и характеризуется нейропатией, атаксией и пигментным ретинитом. Такое разнообразное распределение митохондриальной ДНК в клетках носит название гетероплазмии.
г) Синдром Кернса-Сейра. Синдром Кернса-Сейра («офтальмоплегия плюс») — спорадическое заболевание, обычно обусловленное крупными перестройками/делециями в митохондриальной ДНК. Болезнь проявляется мозжечковой атаксией, прогрессирующим парезом всех глазодвигательных мышц до офтальмоплегии, пигментной ретинопатией и нарушениями в проводящей системе сердца. При патоморфологическом исследовании выявляется губкообразная дегенерация серого и белого вещества с гибелью нейронов, происходящей преимущественно в мозжечке.
В основе заболевания лежат крупные нарушения в митохондриальном геноме.
д) Болезнь Альперса. Для этого заболевания характерно сочетание неврологической симптоматики с признаками дисфункции печени и патоморфологическими признаками гепатита и пролиферации желчных протоков. Болезнь Альперса обычно проявляется в первые годы жизни тяжелыми резистентными судорогами, затем отмечаются задержка развития, гипотонус, атаксия и кортикальная слепота. Характерны гибель нейронов в коре больших полушарий и глубинных структурах, а также губкообразная дегенерация серого вещества. При болезни Альперса были обнаружены мутации ядерного гена, кодирующего у-изоформу ДНК-полимеразы, которая отвечает за репликацию генома митохондрий.
- Рекомендуем ознакомиться со следующей статьей "Механизм поражения нервной системы при дефиците витаминов"
Оглавление темы "Патология нервной системы":- Механизм развития (патогенез) спиноцеребеллярных атаксий
- Механизм развития (патогенез) атаксии Фридрейха
- Механизм развития (патогенез) атаксии-телеангиэктазии
- Механизм развития (патогенез) бокового амиотрофического склероза (БАС)
- Механизм развития (патогенез) бульбоспинальной атрофии (синдрома Кеннеди)
- Классификация генетических метаболических болезней нервной системы
- Механизм развития (патогенез) нейрональных болезней накопления
- Механизм развития (патогенез) лейкодистрофий
- Механизм развития (патогенез) митохондриальной энцефаломиопатии
- Механизм поражения нервной системы при дефиците витаминов