
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) миелодиспластического синдрома
Миелодиспластическими синдромами называют группу клональных нарушений стволовых клеток, характеризующихся дефектами, ассоциированными с неэффективным гемопоэзом и высоким риском трансформации в ОМЛ. При МДС костный мозг частично или полностью замещается клональными потомками неопластической стволовой клетки, которые сохраняют способность к дифференцировке, однако дифференцировка неэффективна или происходит беспорядочно. Эти аномальные клетки остаются в костном мозге, и у пациента в периферической крови определяется цитопения.
Миелодиспластические синдромы (МДС) могут быть первичными (идиопатическими) или вторичными, которые развиваются после химио- или радиотерапии (т-МДС). т-МДС обычно возникают через 2-8 лет после токсического воздействия. В ОМЛ могут перейти все виды МДС, однако наиболее часто и быстро такая трансформация происходит с т-МДС. Хотя в костном мозге и периферической крови обычно обнаруживаются характерные морфологические изменения, для диагностики необходимы и другие лабораторные данные. Особенно полезен цитогенетический анализ, т.к. часто наблюдаются определенные хромосомные аберрации.
а) Молекулярный патогенез. Патогенез МДС изучен недостаточно. При МДС костномозговые клетки-предшественники подвергаются апоптозу в увеличенном объеме, что служит отличительным признаком неэффективного гемопоэза. В связи с этим трудно понять, каким образом при МДС клетки-предшественники получают избирательное преимущество над сохранившимися нормальными клетками-предшествен-никами в костном мозге. Предполагают, что опухоль возникает после повреждения или истощения стволовых клеток.
И первичные, и вторичные МДС ассоциируются с такими хромосомными аномалиями, как моносомии по 5-й и 7-й хромосомам, делеции 5q, 7q и 20q и трисомия по 8-й хромосоме.
б) Морфология миелодиспластического синдрома. Ко времени постановки диагноза в костном мозге обычно отмечают гиперклеточность, но иногда костный мозг является нормоклеточным или, реже, гипоклеточным. Характерны нарушения дифференцировки клеток-предшественников эритроцитов, гранулоцитов, моноцитов и мегакариоцитов, в частности миелодиспластический синдром.
Наиболее типичные нарушения клеток-предшественников эритроцитов: присутствие кольцевидных сидеробластов, эритробластов с нагруженными железом митохондриями, которые имеют вид перинуклеарных гранул в аспиратах или биоптатах, окрашенных берлинской лазурью; мегалобластное созревание, напоминающее таковое при дефиците витамина В12 и фолатов; аномалии ядерного почкования, проявляющиеся в виде деформированных, часто полиплоидных ядер.
Нейтрофилы содержат сниженное количество вторичных гранул, токсичные гранулы и/или тельца Деле. Как правило, наблюдаются псевдоклетки Пельгера-Хюэта (нейтрофилы с двумя ядерными сегментами), иногда встречаются нейтрофилы с полным отсутствием сегментации ядра. Характерны также мегакариоциты с однодольчатым ядром или множественными отдельными ядрами.
Количество миелобластов может быть повышено, однако они составляют < 20% общего количества клеток костного мозга. Кровь часто содержит псевдоклетки Пельгера-Хюэта, гигантские тромбоциты, макроциты и пойкилоциты на фоне относительного или абсолютного моноцитоза. Миелобласты обычно составляют < 10% лейкоцитов крови.
в) Клиническое течение. Первичные МДС — это заболевание преимущественно лиц пожилого возраста; средний возраст составляет 70 лет. До 50% МДС обнаруживают случайно, при стандартном анализе крови. Проявления МДС: слабость, инфекции и геморрагии, обусловленные панцитопенией.
В классификации ВОЗ первичные миелодиспластические синдромы разделены на 5 морфологических типов (их детальное описание не входит в нашу задачу). Типы с более высокой пропорцией бластов ассоциируются с выраженной цитопенией, повышенным риском перехода в ОМЛ и плохим прогнозом. Наличие множественных клональных хромосомных аномалий и степень цитопении — независимые факторы риска, предвещающие неблагоприятный исход.
Медиана выживаемости при первичных миелодиспластических синдромов варьирует от 9 до 29 мес, но некоторые больные из группы с благоприятным прогнозом живут 5 лет и больше. Трансформация в ОМ Л происходит у 10-40% пациентов и обычно сопровождается дополнительными цитогенетическими аномалиями. Пациенты часто умирают от кровотечения в результате тромбоцитопении и инфекции вследствие нейтропении.
Прогноз хуже в случае т-МДС: медиана выживаемости составляет всего 4-8 мес. Цитопения при т-МДС обычно более тяжелая, и переход в ОМЛ часто происходит быстро. Выбор методов лечения МДС довольно ограничен. У молодых пациентов трансплантация аллогенного костного мозга дает шанс на восстановление гемопоэза и продолжительную выживаемость. Пожилые больные получают поддерживающую терапию (антибиотики и гемотрансфузии).
Лекарственные средства, подобные талидомиду (по-видимому, влияющие на взаимодействие клеток-предшественников со стромальными клетками костного мозга), и ингибиторы ДНК-метилазы повышают эффективность гемопоэза и количество форменных элементов крови у некоторых пациентов.
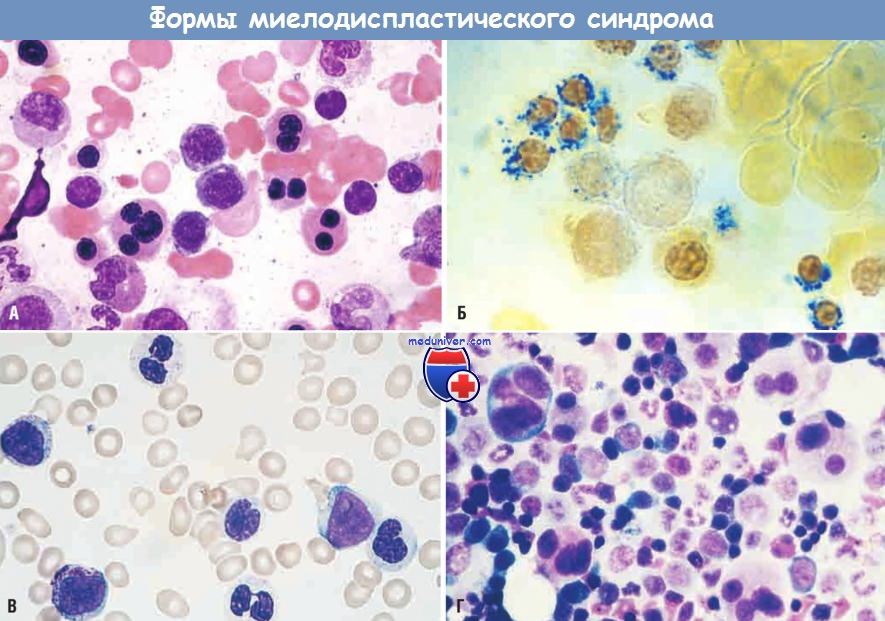
(А) Ядросодержащие клетки-предшественники эритроцитов с многодольчатыми или множественными ядрами (аспират костного мозга).
(Б) Кольцевидные сидеробласты — эритроидные клетки-предшественники с митохондриями, нагруженными железом и имеющими вид синих перинуклеарных гранул
(окрашивание берлинской лазурью, аспират костного мозга).
(В) Псевдоклетки Пельгера-Хюэта (внизу и вверху), представляющие собой нейтрофилы,
ядро которых состоит только из 2 сегментов вместо 3-4, как это должно быть в норме (мазок периферической крови).
(Г) Мегакариоциты с множественными ядрами вместо одиночного многодольчатого ядра в норме (аспират костного мозга).
- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) миелопролиферативного заболевания"
Оглавление темы "Патогенез болезней крови":- Механизмы развития (патогенез) миелодиспластического синдрома
- Механизмы развития (патогенез) миелопролиферативного заболевания
- Механизмы развития (патогенез) хронической миелоидной лейкемии
- Механизмы развития (патогенез) истинной полицитемии
- Механизмы развития (патогенез) эссенциального тромбоцитоза
- Механизмы развития (патогенез) первичного миелофиброза
- Механизмы развития (патогенез) гистиоцитоза клеток Лангерганса
- Строение и физиология селезенки
- Классификация причин спленомегалии