
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) лейкодистрофий
а) Болезнь Краббе. Эта лейкодистрофия с наследованием по аутосомно-рецессивному типу возникает вследствие дефицита галактоцереброзида-галактозидазы (галактозилцерамидазы) — фермента, участвующего в расщеплении галактоцереброзида до церамида и галактозы. Всего известно более 40 мутаций гена, кодирующего этот белок и расположенного на хромосоме 14q31.
Накопление галактоцереброзида само по себе не оказывает токсического воздействия при этом заболевании. Считается, что остаток жирной кислоты отщепляется от этой молекулы за счет альтернативного пути катаболизма, в результате образуется галактосфингозин, который обладает цитотоксическим действием и может поражать олигодендроциты.
Заболевание манифестирует обычно между 3 и 6 мес жизни, быстро прогрессирует и редко продолжается более 2 лет. В симптоматике преобладают различные двигательные расстройства, включая ригидность и слабость мышц. Постепенно возникают трудности с пероральным кормлением. В ЦНС и в периферических нервах выявляются демиелинизация и деструкция олигодендроцитов, нейроны и аксоны относительно сохранны.
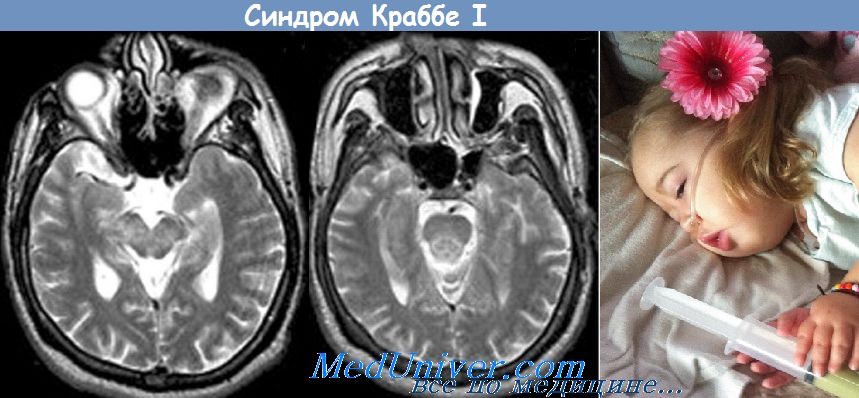
Уникальным диагностически значимым признаком болезни Краббе является агрегация набухших макрофагов (шаровидные клетки) в паренхиме и периваскулярно. Возможно лечение заболевания путем трансплантации пуповинной крови на досимптомной стадии.
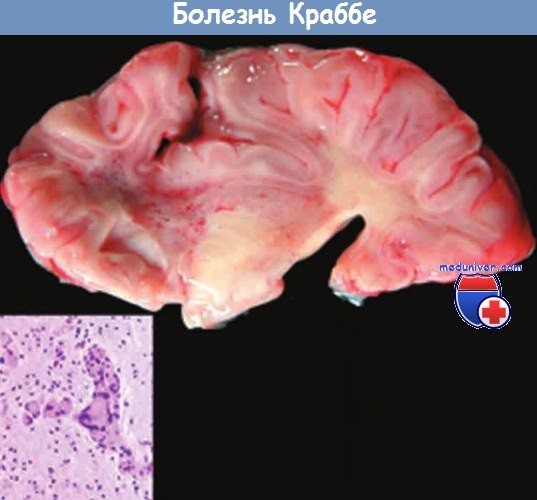
Врезка: шаровидные клетки — «визитная карточка» заболевания.
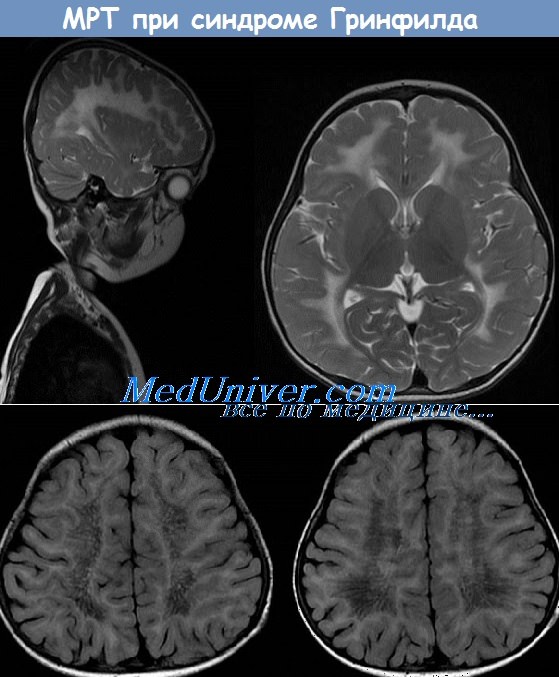
б) Метахроматическая лейкодистрофия (синдром Гринфилда). Это заболевание наследуется по аутосомно-рецессивному типу и характеризуется дефицитом лизосомного фермента арилсульфатазы А. Этот фермент отсоединяет сульфат от сульфатсодержащих липидов (сульфатидов), осуществляя первый этап их утилизации. Дефицит этого фермента приводит к накоплению сульфатидов, в основном цереброзидсульфата, и распаду миелина (каким образом дефицит фермента приводит к распаду миелина, неизвестно, хотя опубликованы данные, что сульфатиды подавляют дифференцировку олигодендроцитов).
Ген, кодирующий арилсульфатазу, находится в дистальном конце хромосомы 22q; известны многочисленные его мутации. Описаны различные клинические виды метахроматической лейкодистрофии: поздняя инфантильная форма (самая частая), ювенильная форма и форма, встречающаяся у взрослых. Первые две манифестируют двигательными нарушениями и прогрессируют медленно (летальный исход наступает через 5-10 лет). У взрослых первыми проявлениями заболевания становятся психические и когнитивные расстройства, а двигательные нарушения присоединяются позднее.
Лечение заключается в трансплантации стволовых клеток костного мозга, но терапия эффективна только до появления неврологических нарушений.
Наиболее важный гистологический признак заболевания — демиелинизация с последующим глиозом. В белом веществе рассеяны макрофаги с вакуолизированной цитоплазмой. Вакуоли содержат сложные кристаллические структуры, состоящие из сульфатидов. Из-за них при окрашивании некоторыми красителями, включая толуидиновый синий, происходит сдвиг спектра поглощения красителя — метахромазия. Похожие изменения наблюдаются и в периферических нервах. Выявление метахроматического субстрата в моче также является чувствительным методом диагностики.
в) Адренолейкодистрофия. Это заболевание имеет несколько генетически и клинически отличающихся разновидностей и характеризуется прогрессирующими симптомами, обусловленными демиелинизацией аксонов центральной и периферической нервной системы, а также надпочечниковой недостаточностью. При раннем появлении симптомов болезнь протекает более быстро. Сцепленное с Х-хромосомой заболевание, как правило, манифестирует в раннем школьном возрасте неврологическими нарушениями и надпочечниковой недостаточностью, протекает быстро и завершается летальным исходом.
При позднем появлении симптомов течение заболевания более медленное. У взрослых на первый план выходят поражения периферических нервов, развивающиеся в течение десятилетий. Заболевание вызвано мутациями гена ALD на хромосоме Xq28, который является членом семейства АТФ-связанных кассетных транспортных белков (ABCD1). Однако корреляции между этими мутациями и течением заболевания почти нет. Болезнь характеризуется невозможностью катаболизма жирных кислот с очень длинной цепью (VLCFA) в пероксисомах, что приводит к увеличению концентрации VLCFA в сыворотке. Происходит демиелинизация в сочетании с глиозом и распространенной лимфоцитарной инфильтрацией. Для заболевания характерна атрофия коры надпочечников, в оставшихся клетках которой определяется накопление VLCFA.
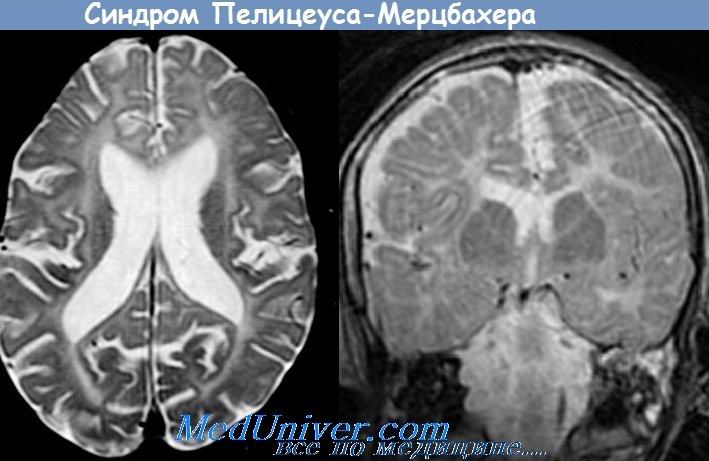
г) Болезнь Пелицеуса-Мерцбахера. Эта сцепленная с Х-хромосомой фатальная лейкодистрофия манифестирует сразу после рождения или в раннем детстве и характеризуется медленно прогрессирующими симптомами поражения белого вещества. У больных детей на начальной стадии заболевания наблюдаются маятникообразные движения глаз, гипотонус, хореоатетоз и пирамидная недостаточность, затем присоединяются спастичность, деменция и атаксия.
Хотя миелин в больших полушариях практически отсутствует, могут определяться отдельные очаги с сохранным миелином, что придает ткани тигроидную окраску при окрашивании на миелин.
Заболевание в большинстве случаев ассоциируется с мутациями гена, расположенного на Х-хромосоме и кодирующего два белка миелина, имеющих альтернативный сплайсинг — протеолипидный белок (PLP) и DM20. Дупликации гена — самая частая мутация; также известны точечные мутации, в результате которых образуются «немые» аллели. До сих пор неизвестно, каким образом эти мутации вызывают заболевание. Тот же локус Х-хромосомы является сайтом мутации, выявляемой при спастической параплегии формы SPG2.
д) Болезнь Канавана. Для этого заболевания характерны мегалэнцефалия, выраженная недостаточность психического развития, слепота и симптомы поражения белого вещества мозга. Симптомы заболевания появляются в раннем детском возрасте и неуклонно прогрессируют, приводя к смерти через несколько лет после манифестации. При аутопсии выявляется губкообразная дегенерация белого вещества. Заболевание обусловлено накоплением N-ацетиласпартата в результате мутаций, приводящих к утрате функции аспартацилазы, кодируемой геном на 17-й хромосоме. Механизм демиелинизации остается неясен.
е) Болезнь Александера. При этом заболевании отмечаются мегалэнцефалия, судорожные приступы и нарастающая задержка психомоторного развития. Морфологически выявляется дегенерация белого вещества, обычно прогрессирующая в лобно-затылочном направлении. Характерными патоморфологическими признаками являются выраженное периваскулярное накопление волокон Розенталя в субпиальных и субэпендимальных областях и в паренхиме мозга. Установлено, что волокна Розенталя состоят из различных белков теплового шока, включая αВ-кристаллин, однако в основе болезни Александера лежат мутации гена, кодирующего белок GFAP. Считается, что заболевание обусловлено мутацией, в результате которой у белка снизилась способность к образованию филаментов и к индукции реакций на стресс.
ж) Лейкодистрофия с исчезновением белого вещества. Лейкодистрофия с исчезновением белого вещества была названа так в связи с особым характером прогрессирования, выявляемого с помощью нейровизуализации. Заболевание вызвано мутациями генов, кодирующих субъединицы фактора инициации трансляции эукариот 2В (eIF2B). Заболевание обычно развивается постепенно в течение первых 20 лет жизни, когда появляются атаксия и судорожные приступы. Болезнь неуклонно прогрессирует и часто осложняется интеркуррентными заболеваниями.
Продолжительность жизни составляет, как правило, несколько лет после манифестации симптомов. Уровни eIF2B в разных участках организма снижены. Механизм избирательного поражения головного мозга с преимущественным повреждением белого вещества неизвестен.
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) митохондриальной энцефаломиопатии"
Оглавление темы "Патология нервной системы":- Механизм развития (патогенез) спиноцеребеллярных атаксий
- Механизм развития (патогенез) атаксии Фридрейха
- Механизм развития (патогенез) атаксии-телеангиэктазии
- Механизм развития (патогенез) бокового амиотрофического склероза (БАС)
- Механизм развития (патогенез) бульбоспинальной атрофии (синдрома Кеннеди)
- Классификация генетических метаболических болезней нервной системы
- Механизм развития (патогенез) нейрональных болезней накопления
- Механизм развития (патогенез) лейкодистрофий
- Механизм развития (патогенез) митохондриальной энцефаломиопатии
- Механизм поражения нервной системы при дефиците витаминов