
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) легочного альвеолярного протеиноза
Легочный альвеолярный протеиноз — это редкое заболевание, которое характеризуется наличием в легких двухсторонних очаговых асимметричных затемнений, выявляемых рентгенологически, что при гистологическом исследовании соответствует накоплению бесклеточного поверхностно-активного вещества внутри просветов альвеол и бронхиол.
Существует 3 варианта легочного альвеолярного протеиноза: приобретенный, врожденный и вторичный, каждый из которых имеет разные механизмы патогенеза и широкий спектр гистологических изменений.
а) Приобретенный легочный альвеолярный протеиноз составляет 90% всех случаев этого заболевания и не имеет наследственной предрасположенности. При исследовании мышей, лишенных гена GM-CSF, неожиданно обнаружили, что эти мыши имели нарушенный клиренс сурфактанта альвеолярными макрофагами, что приводило к состоянию, напоминающему легочный альвеолярный протеиноз у человека.
Впоследствии были найдены ГМ-КСФ-нейтрализующие аутоантитела, циркулирующие в сыворотке крови и бронхиальной слизи пациентов с приобретенным легочным альвеолярным протеинозом. Такие аутоантитела при врожденном или вторичном легочном альвеолярном протеинозе отсутствуют. В настоящее время считают, что за развитие заболевания отвечают антитела к ГМ-КСФ. Эти антитела ингибируют активность эндогенного ГМ-КСФ, что приводит к функциональной недостаточности ГМ-КСФ.
Системность синтеза антител позволила понять патогенез рецидива легочного альвеолярного протеиноза после двухсторонней трансплантации легких. Таким образом, приобретенный легочный альвеолярный протеиноз является аутоиммунным заболеванием.
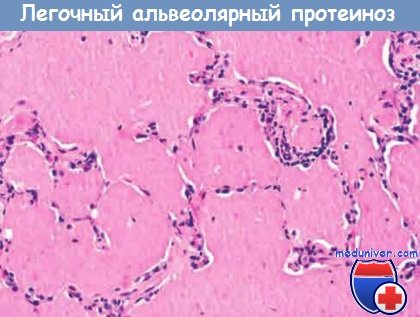
Альвеолы заполнены плотными аморфными белково-жировыми преципитатами; альвеолярные стенки не изменены.
б) Врожденный легочный альвеолярный протеиноз — редкая причина острого респираторного дистресс-синдрома новорожденных. Выявлены мутации нескольких генов, включая ABCA3, кодирующий белок А3 семейства АТФ-связывающих кассетных транспортных белков, гены белков сурфактанта В (SP-B) и белков сурфактанта С (SP-C), ГМ-рецептора общей b-цепи ГМ-КСФ, IL-3, IL-5. Белок АВСАЗ, вероятно, участвует в транспорте компонентов сурфактанта.
Дефицит SP-B наследуется по аутосомно-рецессивному типу и чаще всего вызван сдвигом рамки считывания при мутации гена SP-B. Это приводит к нестабильности мРНК SP-B, снижению или отсутствию SP-B, вторичным нарушениям SP-C и накоплению SP-A и SP-C внутри альвеол.
в) Вторичный легочный альвеолярный протеиноз наблюдается редко. Основные причины данного заболевания — гемопоэтические расстройства, злокачественные опухоли, иммунодефицит, непереносимость белка лизина, острый силикоз и другие респираторные синдромы.
Морфология. Заболевание характеризуется наличием однородного зернистого преципитата в альвеолах с минимальной воспалительной реакцией в виде очаговосливных фокусов в легких с большой площадью поражения. Данные изменения приводят к заметному увеличению размеров и массы легкого. Во время аутопсии с поверхности разреза легочной ткани в этих участках стекает мутная жидкость.
Альвеолярный преципитат PAS-положительный, содержит кристаллы холестерина. Иммуногистохимически выявляются SP-A и SP-C. При врожденном легочном альвеолярном протеинозе обнаруживается дефицит только SP-B, а при приобретенном — всех трех белков сурфактанта. При ультраструктурном исследовании выявляют нарушение образования пластинчатых телец в пневмоцитах II типа, что сочетается с мутациями генов SP-B, SP-C и АВСАЗ.
У взрослых пациентов в основном отмечают неспецифическое нарушение дыхания, которое постепенно прогрессирует и проявляется продуктивным кашлем с обильной мокротой, часто имеющей желеобразные включения. В некоторых случаях описанные симптомы наблюдаются годами, нередко сопровождаются лихорадкой. Эти пациенты составляют группу риска по развитию вторичных инфекций, вызванных различными микроорганизмами.
Возможно прогрессирование одышки, цианоза и дыхательной недостаточности, но иногда заболевание имеет доброкачественное течение с полным выздоровлением больного. Тотальный лаваж легких остается действенным современным стандартом медицинской помощи при легочном альвеолярном протеинозе, в то время как терапия ГМ-КСФ эффективна у 50% пациентов.
Врожденный легочный альвеолярный протеиноз у новорожденных является фатальным респираторным заболеванием. Как правило, младенец доношенный, но вскоре после рождения у него быстро развивается и прогрессирует респираторный дистресс-синдром. Без трансплантации легких летальный исход наступает между 3 и 6 мес жизни.
- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) ТЭЛА"
Оглавление темы "Патогенез болезней легких":- Механизмы развития (патогенез) легочной эозинофилии
- Механизмы развития (патогенез) интерстициальной болезни легких связанной с курением
- Механизмы развития (патогенез) легочного альвеолярного протеиноза
- Механизмы развития (патогенез) ТЭЛА
- Механизмы развития (патогенез) легочной гипертензии
- Механизмы развития (патогенез) синдрома Гудпасчера
- Механизмы развития (патогенез) идиопатического гемосидероза легких
- Механизмы развития (патогенез) легочных инфекций
- Механизмы развития (патогенез) внебольничной пневмонии
- Механизмы развития (патогенез) атипичной пневмонии