
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизмы развития (патогенез) легочной гипертензии
Периферическое сосудистое сопротивление в легких невысоко, поэтому легочное артериальное давление составляет лишь 12-13% от системного артериального давления. Диагноз «легочная гипертензия» ставят, когда среднее легочное давление достигает 25% от системного артериального давления. Клиническая классификация ЛГ основана на общих характеристиках патофизиологических механизмов, клинических проявлениях и терапевтических возможностях:
(1) артериальная ЛГ;
(2) ЛГ в сочетании с болезнями левых отделов сердца;
(3) ЛГ, связанная с заболеваниями легких и/или гипоксией;
(4) ЛГ вследствие хронического тромбоза и/или ТЭЛА;
(5) другие варианты ЛГ.
Легочная гипертензия (ЛГ) наиболее часто связана со структурными перестройками в сердечно-легочной системе, при которых увеличиваются объем и/или давление легочного кровотока, повышается легочное сосудистое сопротивление или возрастает сопротивление кровотока в левых отделах сердца. Эти перестройки являются следствием следующих заболеваний:
- хронической обструктивной болезни легких. У пациентов с этим заболеванием отмечаются гипоксия и разрушение легочной паренхимы, следовательно, уменьшается альвеолярный кровоток. Это приводит к увеличению артериального легочного сопротивления, а затем к повышению давления в легочной артерии;
- врожденных или приобретенных болезней сердца. ЛГ возникает у пациентов с митральным стенозом, например из-за увеличения давления в левом предсердии, что повышает легочное венозное давление и, следовательно, увеличивает давление в легочной артерии;
- рецидивирующей ТЭЛА. Пациенты с рецидивирующей ТЭЛА могут иметь ЛГ в основном из-за уменьшения функциональной площади легочного сосудистого русла, вызванного обтурирующими эмболами, что, в свою очередь, приводит к увеличению легочного сосудистого сопротивления;
- заболеваний соединительной ткани. Некоторые из этих заболеваний (особенно системный склероз) поражают артерии легких, что приводит к воспалению, фиброзу интимы, гипертрофии медии и ЛГ;
- синдрома обструктивного апноэ во сне. Это распространенное заболевание ассоциируется с ожирением и, как установлено в настоящее время, вносит значительный вклад в развитие ЛГ и легочного сердца.
В редких случаях ЛГ развивается спорадически. У таких пациентов все известные причины повышенного давления в легочной артерии исключены. Это заболевание называют идиопатической легочной гипертензией. Еще более редким заболеванием является семейная легочная гипертензия с аутосомно-доминантным типом наследования. В этих семьях присутствует неполная пенетрантность генов, но только в 10-20% случаев наблюдается развернутая клиническая картина болезни.
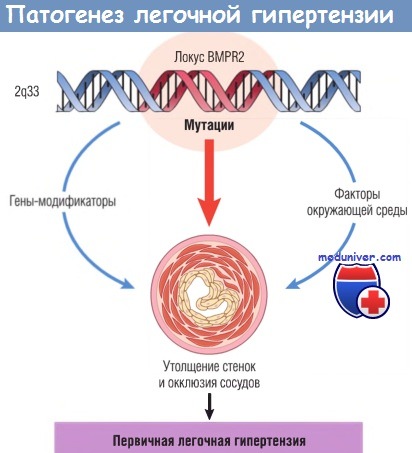
а) Патогенез. Как это часто бывает, многое о патогенезе ЛГ узнали, исследуя молекулярную основу семейной ЛГ.
Эти исследования показали, что семейная ЛГ обусловлена мутациями гена BMPR2, кодирующего рецептор костного морфогенетического белка (BMP) 2-го типа. Чтобы понять, как такие мутации приводят к ЛГ, рассмотрим сосудистую патологию при ЛГ и оценим физиологическую роль сигнального пути BMPR2.
Легочная гипертензия (ЛГ) обусловлена обструкцией сосудов, вызванной пролиферацией эндотелиальных, гладкомышечных и других клеток интимы, что сопровождается концентрическим фиброзом интимы. Как BMPR2 приводит к этим изменениям?
BMPR2 — поверхностный клеточный белок, рецептор суперсемейства TGF-p. BMPR2 связывается с различными цитокинами, в т.ч. TGF-b, BMP, активином и ингибином. Первоначально сигнальный путь был описан в контексте роста костей, однако теперь стало известно, что взаимодействие BMP и BMPR2 играет важную роль в эмбриогенезе, апоптозе, пролиферации и дифференцировке клеток. Конкретные эффекты зависят от вида ткани и ее микросреды. В сосудистых гладкомышечных клетках сигнал BMPR2 вызывает торможение пролиферации и способствует апоптозу.
Таким образом, при отсутствии такого сигнала можно ожидать увеличение выживаемости гладкомышечных клеток и их пролиферацию. В соответствии с этим инактивирующие герминогенные мутации гена BMPR2 находят в 50% случаев семейной ЛГ и в 25% случаев спорадической ЛГ. Во многих семьях, даже без мутаций в кодирующих областях гена BMPR2, обнаружена связь с локусом BMPR2 на хромосоме 2q33, что указывает на другие повреждения гена, такие как перестройки, делеции и инсерции.
Некоторые вопросы остаются без ответа. Во-первых, каким образом утрата одного аллеля гена BMPR2 приводит к отсутствию его сигнала? Существует два возможных объяснения: мутация может действовать как доминантный негативный фактор или происходит утрата второго нормального аллеля, что приводит к утрате гомозиготности BMPR2. Это напоминает герминогенные мутации генов-супрессоров опухолей, приводящие к развитию неоплазий. Интересно, что в некоторых исследованиях сосудистых поражений наблюдали микросателлитную нестабильность в пролиферирующих эндотелиальных клетках.
Это также может приводить к утрате нормального аллеля в клетках сосудистой стенки. Обратите внимание, что подобный механизм инактивирует рецепторы TGF-b в случае наследственного неполипозного рака толстой кишки. Второй вопрос, не имеющий ответа: почему болезнь фенотипически проявляется только у 10-20% лиц с мутациями BMPR2? Это указывает на существование генов-модификаторов и/или влияние факторов окружающей среды. Среди генов-модификаторов есть те, которые контролируют сосудистый тонус, в т.ч. гены простагландина I2, эндотелинсинтетазы и ингибиторов ангиотензинпревращающего фермента. Какие факторы окружающей среды оказывают влияние на развитие ЛГ, остается неизвестным, вероятно, они вызывают дисфункцию вазорегуляторных механизмов. Была предложена двойная модель патогенеза, однако для развития болезни у людей с мутациями BMPR2 должно быть сочетание генетических и внешних факторов.
При вторичной ЛГ возникает дисфункция эндотелия, которую инициируют повреждение эндотелия в артериовенозных шунтах, а также биохимические повреждения, вызванные отложениями фибрина при тромбоэмболии. Уменьшение выработки простагландина I2, снижение синтеза оксида азота и увеличение секреции эндотелина могут способствовать вазоконстрикции ветвей легочной артерии. Кроме того, уменьшение выработки простагландина I2 и оксида азота обусловливает адгезию тромбоцитов и их активацию.
В результате активации эндотелиальные клетки становятся тромбогенными, что приводит к постоянному отложению фибрина. Наконец, синтез и секреция факторов роста и цитокинов индуцируют миграцию и репликацию гладкомышечных клеток и выработку ВКМ.
В патогенезе ЛГ у некоторых людей присутствует вазоспастический компонент. У таких больных после приема вазодилататоров периферическое легочное сосудистое сопротивление быстро снижается. Тем не менее есть сообщения о развитии ЛГ после применения тропического растения Crotalaria spectabilis (используют в медицине в качестве добавки в чай), препарата аминорекса (регулятора аппетита), фальсифицированного оливкового масла, препаратов для похудения фенфлурамина и фентермина. Предположительно такие вещества действуют опосредованно (повышая экспрессию или активность транспортера серотонина).
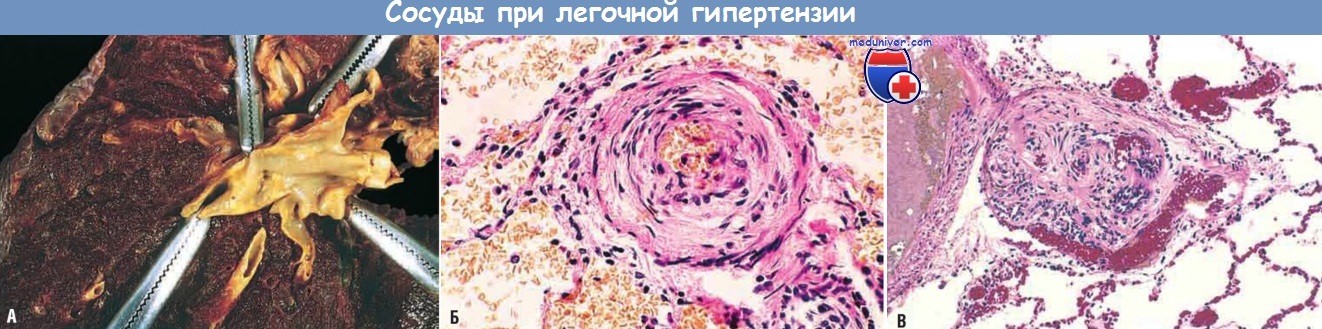
(А) Макроскопический вид атеросклеротической бляшки (обычно располагаются в крупных сосудах).
(Б) Выраженная гипертрофия медии.
(В) Плексиформные структуры, характерные для поражения мелких артерий при прогрессирующей легочной гипертензии.
б) Морфология. Некоторые патологические характеристики ЛГ присущи всем ее формам независимо от этиологии, например гипертрофия медии артерий мышечного и эластического типа, атеросклероз легочной артерии и ее основных ветвей, гипертрофия правого желудочка. Наличие множества организованных или реканализированных тромбов свидетельствует о рецидивирующей ТЭЛА как о причине ЛГ, а сочетание тромбов с диффузным фиброзом легких, тяжелой эмфиземой или хроническим бронхитом указывает на наличие у пациента хронической гипоксии как инициирующего события тромбоэмболии. Сосудистые изменения могут касаться всего артериального дерева — от основных ветвей легочных артерий до артериол. В наиболее тяжелых случаях атеросклеротические бляшки образуются в легочной артерии и ее основных ветвях, как при системном атеросклерозе (но выражены в меньшей степени).
Артериолы и мелкие артерии (40-300 мкм в диаметре) поражаются больше всех, что проявляется резким увеличением толщины их мышечной оболочки (медиальной гипертрофией) и фиброзом интимы. Иногда сужение просвета артериол и мелких артерий становится критическим. Наиболее тяжелые изменения в виде плексиформных структур обнаруживаются при идиопатической и семейной ЛГ, врожденных пороках сердца без лечения с формированием артериовенозных шунтов, а также при ЛГ, ассоциированной со злоупотреблением наркотическими средствами и ВИЧ-инфекцией. Плексиформные структуры состоят из пучков капилляров, формирующих сеть, которая окружает просветы расширенных и тонкостенных мелких артерий. Также могут присутствовать расширенные сосуды и признаки артериита. В отдельных случаях причиной ЛГ может быть обширная окклюзия вен фиброзной тканью .
в) Клинические признаки. Первичную (идиопатическую) ЛГ наиболее часто выявляют у женщин в возрасте 20-40 лет и иногда у детей младшего возраста.
Клинические признаки и симптомы всех форм легочной гипертензии (ЛГ) становятся очевидными только на поздних стадиях. В случаях первичной ЛГ основными клиническими симптомами являются одышка и утомляемость, а у некоторых пациентов — боль в груди по типу стенокардии. Со временем развиваются тяжелая дыхательная недостаточность, цианоз, гипертрофия правого желудочка. Летальный исход, как правило, наступает через 2-5 лет (80%) от правожелудочковой недостаточности (декомпенсация легочного сердца), часто в сочетании с ТЭЛА и пневмонией.
Обычные методы лечения (кислородная поддержка, блокаторы кальциевых каналов, антикоагулянты, дигоксин и диуретики) эффективны при краткосрочных курсах терапии. Однако разработанные в последние годы методы лечения, в частности введение аналогов простагландина I2, антагонистов эндотелиальных рецепторов, вдыхание монооксида азота и использование ингибиторов фосфодиэстеразы-5, позволили улучшить результаты терапии у многих пациентов. У некоторых пациентов возникает необходимость в трансплантации легких. Генная терапия, успешная в экспериментах на животных, в будущем станет доступной и для человека.
- Рекомендуем ознакомиться со следующей статьей "Механизмы развития (патогенез) синдрома Гудпасчера"
Оглавление темы "Патогенез болезней легких":- Механизмы развития (патогенез) легочной эозинофилии
- Механизмы развития (патогенез) интерстициальной болезни легких связанной с курением
- Механизмы развития (патогенез) легочного альвеолярного протеиноза
- Механизмы развития (патогенез) ТЭЛА
- Механизмы развития (патогенез) легочной гипертензии
- Механизмы развития (патогенез) синдрома Гудпасчера
- Механизмы развития (патогенез) идиопатического гемосидероза легких
- Механизмы развития (патогенез) легочных инфекций
- Механизмы развития (патогенез) внебольничной пневмонии
- Механизмы развития (патогенез) атипичной пневмонии