
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) гемохроматоза
Гемохроматоз впервые был описан Ф. Реклингаузеном в 1889 г. Заболевание характеризуется выраженным накоплением железа, большая часть которого откладывается в паренхиматозных органах — печени и поджелудочной железе. Железо также может накапливаться в сердце, суставах или эндокринных органах.
Первичный, или наследственный, гемохроматоз — гомозиготное рецессивное наследственное заболевание, вызванное повышенным всасыванием железа. Вторичный, или приобретенный, гемохроматоз — это накопление железа в тканях в результате парентерального введения препаратов железа (обычно в форме трансфузий) или по другим причинам. Мы будем использовать термин «гемохроматоз» для обозначения наследственного заболевания, а термин «гемосидероз» — для приобретенного.
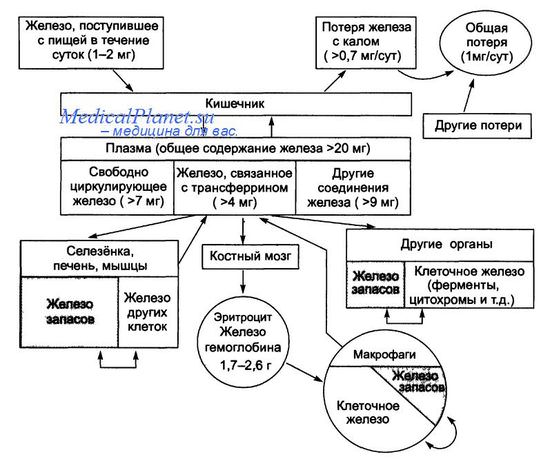
Общий пул железа в организме взрослого человека в норме варьирует от 2 до 6 г. Приблизительно 0,5 г железа аккумулируется в печени, а 98% из этого количества накапливается в гепатоцитах. При гемохроматозе общее количество железа в организме может достигать 50 г, при этом более 30% его накапливается в печени. Для гемохроматоза характерны следующие признаки:
- железо накапливается в течение всей жизни, а повреждения, вызываемые повышенным содержанием железа, прогрессируют медленно, поэтому заболевание обычно манифестирует в возрасте 40-60 лет;
- среди больных преобладают мужчины (соотношение мужчин и женщин составляет 5-7:1). Клинические симптомы заболевания у мужчин проявляются значительно раньше. Это частично связано с тем, что у женщин накопление железа происходит медленнее из-за его физиологической потери (менструации, беременность);
- при развернутой картине заболевания мелкоузловой цирроз печени выявляют у всех пациентов, сахарный диабет — у 75-80% больных, гиперпигментацию кожи — у 75-80% пациентов.

а) Патогенез. Поскольку в организме человека отсутствуют механизмы регуляции выведения железа, уровень его полностью зависит от интенсивности всасывания в кишечнике. При гемохроматозе всасывание железа, поступающего с пищей, аномально возрастает, что приводит к постепенному накоплению железа (0,5-1,0 г в год) преимущественно в печени. Болезнь проявляется клинически после накопления в организме 20 г железа. Высокая концентрация железа оказывает прямое токсическое воздействие на ткани организма за счет:
(1) перекисного окисления липидов под действием свободных радикалов, катализируемых ионами железа;
(2) стимуляции образования коллагена путем активации звездчатых клеток печени;
(3) взаимодействия АФК и железа с ДНК, что приводит к необратимому повреждению клетки или предрасполагает к развитию ГЦК.
При обратимом повреждении клеток удаление избытка железа в ходе лечения способствует восстановлению функции пораженной ткани.
Основным регулятором всасывания железа является белок гепсидин (пептид, экспрессируемый в печени, LEAP1), кодируемый геном НАМР и обладающий антимикробной активностью. Гепсидин вырабатывают гепатоциты в виде пропептида, состоящего из 84 аминокислот, затем он расщепляется на активный белок, содержащий 25 аминокислот, и мелкие циркулирующие полипептиды, состоящие из 20 и 23 аминокислот. Транскрипция гепсидина усиливается под влиянием провоспалительных цитокинов и железа, а снижается при дефиците железа, гипоксии и неэффективном эритропоэзе. Гепсидин связывается с белком клеточного канала выведения железа ферропортином, вызывая его интернализацию и протеолиз, что препятствует высвобождению железа из макрофагов и клеток тонкой кишки. Так гепсидин снижает уровень железа в плазме крови. Таким образом, дефицит гепсидина приводит к повышению концентрации железа в плазме.
Регулируют содержание гепсидина и другие белки, участвующие в метаболизме железа, а именно:
(1) гемоювелин (HJV), который экспрессируется в печени, сердце и скелетных мышцах;
(2) рецептор трансферрина 2-го типа (TfR2), который в большой концентрации содержится в гепатоцитах и способствует поглощению связанного с трансферрином железа;
(3) белок HFE, продукт гена гемохроматоза.
Отсутствие экспрессии гепсидина из-за мутации его гена, а также генов HJV, TfR2 и HFE приводит к развитию гемохроматоза. Среди этих мутаций самыми частыми являются мутации гена HFE. Мутации генов НАМР и HJV приводят к развитию тяжелого наследственного гемохроматоза, известного как ювенильный гемохроматоз. Мутации генов HFE и TfR2 являются причиной классического наследственного гемохроматоза взрослых, более легкого заболевания по сравнению с ювенильным гемохроматозом. Мутации гена FPN вызывают болезнь накопления железа. Точный механизм, посредством которого белки HFE, HJV и TfR2 регулируют активность гепсидина и ферропортина, еще предстоит выяснить. Недавно было установлено, что сериновая протеаза (TMPRSS6) является рецептором железа и подавляет экспрессию НАМР.
Гемосидероз взрослых практически всегда ассоциируется с мутацией гена HFE, который локализуется на коротком плече 6-й хромосомы в локусе 6р21.3, вблизи локуса гена HLA. Ген HFE кодирует синтез веществ, подобных молекулам HLA класса I, которые регулируют всасывание в тонкой кишке поступающего с пищей железа. Самой частой мутацией гена HFE является замена аминокислоты цистеин на тирозин в позиции 282 (C282Y) вследствие простой замены гуанина (G) на аденин (А) в нуклеотидной последовательности 845 (G845A). Такая мутация инактивирует белок и присутствует у 70-100% пациентов с диагнозом «наследственный гемохроматоз». Еще одной частой мутацией является H63D (замещение гистидина в позиции 63 на аспартат). У гомозигот с H63D и гетерозигот со смешанной мутацией C282Y/H63D отмечается лишь незначительное накопление железа.
Мутация C282Y обычно определяется у представителей европеоидной расы, тогда как мутация H63D распространена повсеместно. Частота гомозиготной мутации C282Y составляет 0,45% (1 на 220 человек), а гетерозиготной — 11%, что делает гемохроматоз одним из наиболее частых наследственных заболеваний человека. Однако пенетрантность этого заболевания очень низка даже у лиц с гомозиготной мутацией C282Y, т.е. даже при наличии генетического дефекта заболевание развивается далеко не у всех.
б) Морфология. Морфологические изменения при наследственном гемохроматозе:
(1) отложение гемосидерина (по мере снижения интенсивности) в печени, поджелудочной железе, миокарде, гипофизе, надпочечниках, щитовидной и паращитовидных железах, суставах и коже (определяется при окрашивании берлинской лазурью или с помощью атомно-абсорбционной спектрометрии);
(2) цирроз печени;
(3) фиброз поджелудочной железы.
В печени на начальных стадиях заболевания железо выявляется в виде желтовато-золотистых гранул гемосидерина в цитоплазме перипортальных гепатоцитов, которые окрашиваются берлинской лазурью в синий цвет. По мере накопления железа отмечаются прогрессирующее вовлечение в процесс остальных гепатоцитов дольки, а также пигментация эпителия протоков и клеток Купфера. Железо обладает прямым гепатотоксическим эффектом, при этом характерно отсутствие воспаления. На этой стадии заболевания печень обычно несколько увеличена в размерах, плотная, цвета темного шоколада. Фиброзные септы образуются медленно, но в конечном итоге в интенсивно пигментированной печени формируется мелкоузловой цирроз.
Стандартом определения уровня железа в печени является биохимический метод. В норме содержание железа в ткани печени составляет менее 1000 мкг на 1 г сухой массы органа. У взрослых с наследственным гемохроматозом концентрация железа в ткани печени превышает 10 тыс. мкг на 1 г сухой массы органа. Содержание железа в ткани печени более 22 тыс. мкг на 1 г сухой массы сопровождается развитием фиброза и цирроза.
Поджелудочная железа становится интенсивно пигментированной, в ней наблюдаются диффузный интерстициальный фиброз, а также атрофия паренхимы. Гемосидерин выявляется в ацинарных клетках и в клетках островков Лангерганса, а иногда и в интерстициальной фиброзной строме. Сердце часто увеличено в размерах, в кардиомиоцитах присутствуют гранулы гемосидерина, придающие миокарду коричневый цвет. Может развиваться незначительный интерстициальный фиброз. Пигментация кожи только частично обусловлена отложением гемосидерина в макрофагах и фибробластах дермы, в основном же пигментация является результатом повышенной продукции меланина в эпидермисе. Комбинация механизмов пигментации обусловливает характерный аспидно-серый цвет кожи. При отложении гемосидерина в синовиальной оболочке суставов может развиться острый синовиит. Избыточное отложение пирофосфата кальция приводит к повреждению суставного хряща и полиартриту (псевдоподагре). Яички могут быть уменьшены в размерах или атрофичны, однако выраженная пигментация отсутствует. В данном случае атрофия является вторичной и возникает вследствие снижения уровня гонадотропина и тестостерона из-за нарушения функции гипоталамо-гипофизарной системы.
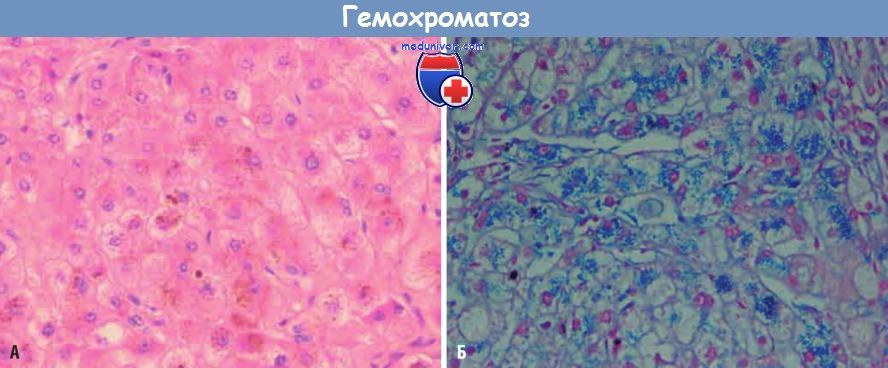
Темнокоричневые гранулы железа в гепатоцитах при окрашивании гематоксилином и эозином (А) и синие при окрашивании берлинской лазурью (Б).
в) Клинические признаки. Классический наследственный гемохроматоз чаще возникает у мужчин и редко манифестирует в возрасте до 40 лет. Основные проявления: гепатомегалия, боль в животе, пигментация кожи (особенно на участках, подвергающихся воздействию солнечных лучей), нарушения гомеостаза глюкозы или сахарный диабет вследствие разрушения островков поджелудочной железы, нарушение функции сердца (аритмия, кардиомиопатия) и атипичный артрит. Некоторые пациенты предъявляют жалобы, обусловленные гипогонадизмом (например, аменорея у женщин, импотенция и снижение либидо у мужчин). Классическая триада симптомов — пигментный цирроз печени с гепатомегалией, пигментация кожи и сахарный диабет — может отсутствовать вплоть до самой поздней стадии заболевания. Смерть может наступить в результате цирроза печени, поражения сердца и ГЦК. Риск развития ГЦК у лиц с гемохроматозом в 200 раз выше, чем в общей популяции, при этом лечение, направленное на снижение концентрации железа, не снижает риска развития опухоли.
К счастью, гемохроматоз легко диагностировать задолго до необратимого повреждения тканей. Для этого используют скрининговые методы: лабораторный анализ для выявления очень высоких концентраций железа и ферритина в сыворотке крови (при исключении других причин накопления железа в организме) и биопсию печени (по показаниям). Важен скрининг членов семьи пробанда. У гетерозигот также отмечается повышенное накопление железа, однако концентрация его не достигает того уровня, при котором происходит выраженное повреждение тканей. В настоящее время у большинства пациентов гемохроматоз диагностируют на субклинической, прецирротической стадии благодаря рутинному исследованию концентрации железа в сыворотке крови. Таким пациентам проводят регулярные кровопускания, что не влияет на продолжительность их жизни.
Гемохроматоз новорожденных (также называемый врожденным гемохроматозом) является заболеванием неизвестной этиологии, проявляющимся тяжелым поражением печени и выраженным внепеченочным отложением гемосидерина. Гемохроматоз новорожденных не относят к наследственным заболеваниям, поскольку повреждение печени у новорожденного, приведшее к накоплению гемосидерина, произошло еще внутриутробно и может быть связано с поражением печени плода иммунной системой матери. Для постановки правильного диагноза необходимо обнаружение внепеченочных отложений гемосидерина в биоптате слизистой оболочки щеки. Проводят симптоматическую терапию, а в тяжелых случаях — трансплантацию печени.
Самыми частыми причинами гемосидероза являются заболевания, сопровождающиеся неэффективным эритропоэзом, в частности тяжелые формы талассемии и миелодиспластические синдромы. При этих заболеваниях избыток железа возникает не только вследствие гемотрансфузий, но также в результате его повышенного всасывания. Сами по себе регулярные переливания крови в течение длительного периода времени (как у пациентов с хронической гемолитической анемией) также могут приводить к развитию системного гемосидероза и повреждению паренхиматозных органов. Алкогольный цирроз печени часто сопровождается умеренным повышением концентрации железа в клетках печени. Этот процесс является этанол-индуцированным перераспределением железа, а общее количество железа в организме возрастает незначительно. Более редкие формы накопления железа, напоминающие наследственный гемохроматоз, в частности сидероз банту, наблюдаются у жителей Африки, проживающих в регионах южнее Сахары. Заболевание развивается в результате употребления в большом количестве алкогольных напитков, брожение которых проходит в железной посуде. Кроме того, в этих популяциях возможны генетические нарушения, например мутации FPN. Наконец, к накоплению железа в гепатоцитах также могут приводить хронические гепатиты В и С.
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) болезни Вильсона-Коновалова"
Оглавление темы "Патогенез заболеваний печени":- Морфология хронического вирусного гепатита
- Механизм развития (патогенез) молниеносного гепатита
- Механизм развития (патогенез) абсцесса печени
- Механизм развития (патогенез) аутоиммунного гепатита
- Механизм развития (патогенез) повреждения печени лекарствами
- Механизм развития (патогенез) повреждения печени алкоголем
- Механизм развития (патогенез) жировой дистрофии печени
- Механизм развития (патогенез) гемохроматоза
- Механизм развития (патогенез) болезни Вильсона-Коновалова
- Механизм развития (патогенез) дефицита альфа-1-антитрипсина