
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) фокального сегментарного гломерулосклероза
Для фокального сегментарного гломерулосклероза почек (ФСГС) характерно поражение некоторых клубочков (отсюда — фокальный), при этом в поврежденном клубочке в процесс вовлечены лишь некоторые петли капилляра (отсюда — сегментарный). Фокальный сегментарный гломерулосклероз (ФСГС) клинически часто проявляется нефротическим синдромом или тяжелой протеинурией.
а) Классификация. Фокальный сегментарный гломерулосклероз (ФСГС) может быть:
- первичным (идиопатический ФСГС);
- сочетающимся с другими состояниями, например ВИЧ (ВИЧ-ассоциированная нефропатия), героиновой зависимостью (героиновая нефропатия), серповидно-клеточной анемией, выраженным ожирением;
- вторичным склеротическим процессом в исходе фокального гломерулонефрита с массивным некрозом (например, нефропатии IgA);
- следствием адаптации на снижение объема ткани почки при прогрессирующей болезни почек (например, рефлюксной и гипертензивной нефропатии) или развиться при односторонней агенезии почек;
- редкой врожденной формой нефротического синдрома, обусловленного мутациями белков щелевой диафрагмы, например подоцина, а-актинина 4 и рецептора потенциал-зависимого кальциевого канала 6 (TRPC6).
Идиопатический Фокальный сегментарный гломерулосклероз (ФСГС), по данным многих ретроспективных исследований, обусловливает 10% случаев нефротического синдрома у детей и 35% — у взрослых. Частота ФСГС (первичного и вторичного) возросла, и на сегодняшний день в США ФСГС является самой распространенной причиной нефротического синдрома у взрослых, особенно среди выходцев из Латинской Америки и лиц с темным цветом кожи.
Клинические проявления фокального сегментарного гломерулосклероза почек (ФСГС) отличаются от таковых при болезни минимальных изменений. При ФСГС:
(1) выше частота гематурии, сниженной СКФ и гипертензии;
(2) протеинурия чаще неселективная;
(3) хуже ответ на кортикостероидную терапию;
(4) идет прогрессирование до хронической нефропатии (как минимум у 50% пациентов в течение 10 лет развивается терминальная стадия хронической болезни почек).
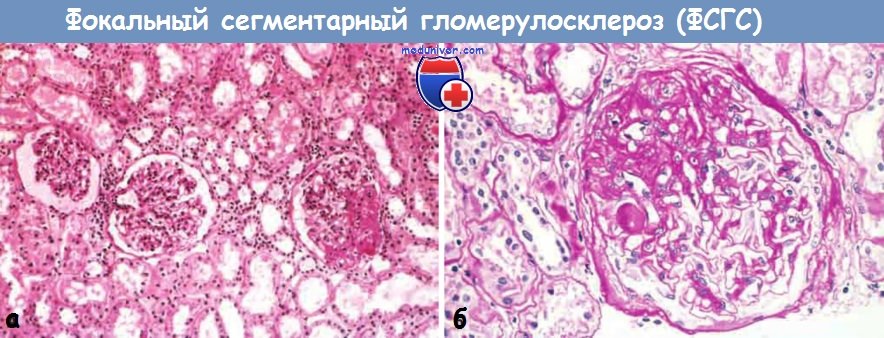
(А) При малом увеличении виден сегментарный склероз одного из трех клубочков (справа).
(Б) При большом увеличении в очагах склероза выявляются отложения гиалина и жировые капли (мелкие вакуоли).
б) Патогенез. В настоящее время нет единого ответа на вопрос: идиопатический ФСГС — это самостоятельная нозологическая единица или стадия развития болезни минимальных изменений? Характерная дегенерация и фокальные разрывы висцерального эпителиального слоя являются результатом диффузных изменений эпителиальных клеток, типичных для болезни минимальных изменений. Такие повреждения эпителия патогномоничны для ФСГС и могут быть обусловлены множеством механизмов, включая циркулирующие цитокины и генетически детерминированные дефекты белков щелевой диафрагмы. Склероз и гиалиноз обусловлены осаждением белков плазмы в очагах крайне высокой проницаемости щелевой диафрагмы и накоплением ВКМ. Рецидив протеинурии у некоторых пациентов (иногда через 24 час после трансплантации почки с последующим явным переходом в ФСГС) подтверждает, что причиной повреждения эпителия является циркулирующий фактор, возможно цитокин. Из сыворотки крови этих больных был выделен вызывающий протеинурию фактор с молекулярной массой 50 кДа, не относящийся к классу Ig, однако более точной информации об этом факторе нет.
в) Морфология. При световой микроскопии выявляют фокальные сегментарные поражения, затрагивающие лишь небольшое количество клубочков. При недостаточном объеме биоптата такие поражения диагностировать трудно. Сначала поражаются клубочки юкстамедуллярной зоны, затем изменения приобретают генерализованный характер. В склерозированных сегментах происходят коллапс петель капилляров, накопление матрикса и сегментарное отложение белков плазмы вдоль стенки капилляра {гиалиноз). Гиалиноз может быть настолько выраженным, что происходит окклюзия просвета капилляра. Часто обнаруживают жировые капли и пенистые клетки. Клубочки, которые не имеют сегментарных поражений, при световой микроскопии обычно выглядят неизмененными, однако возможно накопление мезангиального матрикса.
При электронной микроскопии выявляют диффузное сглаживание малых отростков подоцитов (в склерозированных и в несклерозированных областях клубочка). При этом происходит очаговое отслоение эпителиальных клеток и оголение базальной мембраны. При иммунофлуоресцентном исследовании в очагах склероза и/или мезангии можно выявить IgM и компонент системы комплемента С3. Помимо фокального сегментарного гломерулосклероза возможны выраженный гиалиноз и утолщение стенок приносящих артериол. При прогрессировании заболевания увеличиваются количество склерозированных клубочков и площадь повреждения. Со временем это приводит к тотальному склерозу клубочков с выраженной атрофией канальцев и интерстициальным фиброзом.
Коллаптоидная гломерулопатия (один из морфологических вариантов ФСГС) характеризуется сморщиванием всего клубочка в сочетании с различными типами ФСГС или без них. Типичными для коллаптоидной гломерулопатии являются пролиферация и гипертрофия висцеральных эпителиальных клеток клубочка. Повреждение может быть идиопатическим, однако наиболее характерно для ВИЧ-ассоциированной нефропатии. В обоих случаях гломерулопатия сопровождается выраженными повреждениями канальцев с формированием микрокист. Прогноз при таком ФСГС чрезвычайно неблагоприятный.
Изучение генетических основ некоторых вариантов ФСГС и других причин нефротического синдрома позволило расширить представления о патогенезе протеинурии и разработать новые методы диагностики и прогнозирования исхода у пациентов с ФСГС. Первым был идентифицирован ген NPHS1, который расположен на хромосоме 19q13 и кодирует белок нефрин. Некоторые мутации гена NPHS1 приводят к развитию врожденного нефротического синдрома финского типа, проявляющегося гломерулопатией, подобной болезни минимальных изменений, с выраженным сглаживанием отростков подоцитов. Анализ гена NPHS1 позволяет пренатально диагностировать врожденный нефротический синдром.
В результате мутаций гена NPHS2, который расположен на хромосоме 1q25-q31 и кодирует синтез белка подоцина, локализующегося в щелевой диафрагме, развивается аутосомно-рецессивный ФСГС. Мутации NPHS2 в ~ 30% случаев обусловливают развитие у детей резистентного к кортикостероидной терапии нефротического синдрома. У больных детей обычно наблюдаются признаки ФСГС.
Мутации гена, кодирующего актинсвязывающий белок а-актинин 4, обусловливают развитие аутосомно-доминантного ФСГС, протекающего вначале без выраженных клинических симптомов, но способного быстро приводить к почечной недостаточности.
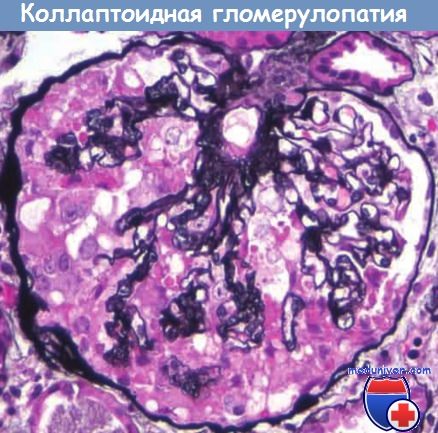
Сморщивание клубочка, сужение просвета капилляров, пролиферация и отек висцерального слоя эпителия.
Подобные изменения обнаруживаются при идиопатической и ВИЧ-ассоциированной нефропатии с помощью метода импрегнации серебром.
Мутации гена, кодирующего TRPC6, были обнаружены у некоторых взрослых единокровных родственников, заболевших ФСГС. TRPC6 секретируется многими клетками, в т.ч. подоцитами. Патогенные мутации нарушают функцию подоцитов из-за повышения поступления кальция в эти клетки.
Объединяет все перечисленные белки то, что все они расположены в щелевой диафрагме и цитоскелете соседних подоцитов. Их специфические функции и механизмы взаимодействия изучены не полностью, но ясно, что целостность каждого из них необходима для обеспечения нормального функционирования фильтрационного барьера клубочка.
Исследования на мышиной модели позволили установить роль еще одного белка — CD2-ассоциированного белка (CD2AP), который редко обнаруживается у человека и может быть связан с развитием протеинурии. До момента идентификации этих генетических нарушений патогенез некоторых случаев идиопатического нефротического синдрома объясняли другими факторами, приводящими к нарушению проницаемости: взаимодействиями между клетками и между клетками и матриксом, особенно теми, которые опосредованы a3b1-интегринами, дистрогликанами и лиминами. Нарушение этих взаимодействий может привести к потере адгезии подоцита к БМК.
Аблационная нефропатия — вторичный ФСГС, развивающийся как осложнение гломерулярных и негломерулярных заболеваний, которые характеризуются снижением массы функционирующей почечной ткани (особенно при рефлюксной нефропатии и односторонней агенезии почки) и могут приводить к гломерулосклерозу и почечной недостаточности. Патогенез был рассмотрен в отдельной статье ранее.
г) Клинические признаки. При идиопатическом ФСГС вероятность спонтанного регресса невелика, а реакция организма на кортикостероидную терапию вариабельная. В целом прогноз у детей лучше, чем у взрослых. Прогрессирование почечной недостаточности происходит с разной скоростью. У 20% больных наблюдаются необычайно быстрое течение болезни с неконтролируемой массивной протеинурией и развитие почечной недостаточности в течение 2 лет. После трансплантации почки рецидив происходит в 25-50% случаев.
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) ВИЧ-ассоциированной нефропатии"
Оглавление темы "Патогенез заболеваний почек":- Механизм развития (патогенез) фокального сегментарного гломерулосклероза
- Механизм развития (патогенез) ВИЧ-ассоциированной нефропатии
- Механизм развития (патогенез) мембранопролиферативного гломерулонефрита (МПГН)
- Механизм развития (патогенез) нефропатии IgA - болезни Берже
- Механизм развития (патогенез) синдрома Альпорта
- Механизм развития (патогенез) болезни тонких базальных мембран
- Механизм развития (патогенез) хронического гломерулонефрита
- Механизм развития (патогенез) поражения почек при пурпуре Шенлейна-Геноха
- Механизм развития (патогенез) поражения почек при эндокардите
- Механизм развития (патогенез) поражения почек при сахарном диабете