
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) дефицита альфа-1-антитрипсина
Дефицит альфа-1-антитрипсина — это аутосомно-рецессивное заболевание, для которого характерна очень низкая концентрация альфа-1-антитрипсина. Основная функция этого белка заключается в подавлении протеаз, в частности эластазы нейтрофилов, катепсина G и про-теиназы 3, которые в норме выделяются нейтрофилами в месте воспаления.
Дефицит альфа-1-антитрипсина приводит к развитию эмфиземы легких в результате активности протеаз. агАнтитрипсин также вызывает заболевания печени вследствие накопления этого белка в гепатоцитах. Кроме того, при дефиците альфа-1-антитрипсина могут наблюдаться кожный панникулит, аневризмы артерий, бронхоэктазы и гра-нулематоз Вегенера.
альфа-1-Антитрипсин — это малый гликопротеин плазмы, который состоит из 394 аминокислот и синтезируется преимущественно гепатоцитами. Он входит в семейство ингибиторов сериновых протеаз (серпины). Ген, расположенный на 14-й хромосоме, обладает высокой степенью полиморфизма.
Идентифицировано по меньшей мере 75 форм альфа-1-антитрипсина. Общее обозначение — Pi (protease inhibitor — ингибитор протеазы), а далее буквами в алфавитном порядке указывают позицию в изоэлектрическом геле. Двумя буквами обозначают генотип двух аллелей. Самый частый генотип — PiMM. Это генотип дикого типа, он присутствует у 90% людей.
Большинство аллелей характеризуются заменами в полипептидной цепи, однако при этом вырабатывается нормальное количество функционально активного альфа-1-антитрипсина. Некоторые дефицитные типы, включая PiS, характеризуются умеренным снижением концентрации альфа-1-антитрипсина в сыворотке крови без клинических проявлений. При редком типе, обозначаемом Pi-нуль, альфа-1-антитрипсин в сыворотке крови вообще не определяется. Наиболее частой клинически значимой мутацией является PiZ.
У гомозигот с генотипом PiZZ уровень циркулирующего альфа-1-антитрипсина составляет лишь 10% нормы. У таких лиц заболевание часто проявляется клинически.
Экспрессия аллелей является аутосомно-кодоминантной, следовательно, у гетерозигот PiMZ — средний уровень альфа-1-антитрипсина в плазме крови. Среди жителей Северной Европы частота генотипа PiS составляет 6%, генотипа PiZ — 4%. Генотип PiZZ выявляют у 1 из 1800 живорожденных детей. Дефицит альфа-1-антитрипсина иногда очень рано проявляется поражением печени и является самой частой генетической причиной заболевания печени у новорожденных и детей.
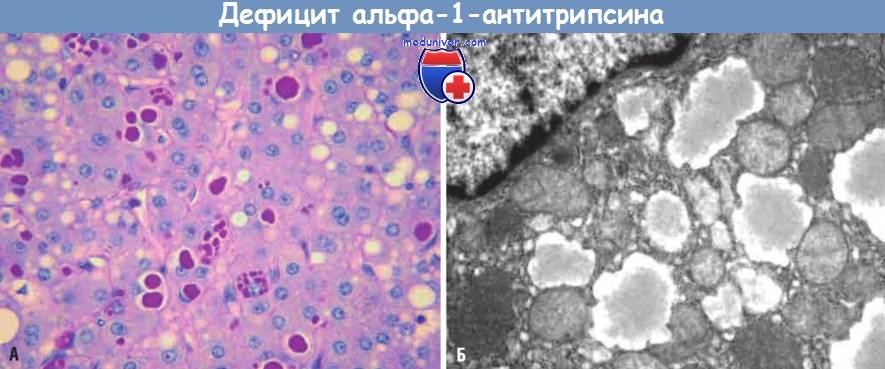
(А) PAS-реакция выявляет характерные красные гранулы в цитоплазме гепатоцитов.
(Б) На электронной микрофотографии показаны расширенные цистерны эндоплазматической сети.
а) Патогенез. При большинстве вариантов аллелей мРНК транскрибируется и -антитрипсин нормально синтезируется и секретируется. При дефицитных типах присутствует избирательный дефект миграции этого белка из эндоплазматической сети в аппарат Гольджи. Это особенно выражено при полипептиде PiZ, для которого характерна замена единственной аминокислоты Glu на Lys в позиции 342.
Мутантный альфа-1-антитрипсин Z (arAT-Z) аномально складывается и полимеризуется, вызывая перегрузку эндоплазматической сети, что приводит к апоптозу клетки. Точный механизм поражения печени белком альфа-1-AT-Z до конца не выяснен. Накопление альфа-1-AT-Z в эндоплазматической сети запускает серию процессов, включая аутофагоцитоз, митохондриальную дисфункцию и, возможно, активацию провоспалительного NF-кВ, что повреждает гепатоциты.
У всех лиц с генотипом PiZZ происходит накопление arAT-Z в эндоплазматической сети гепатоцитов, но только у 10-15% из них развивается заболевание.
Считается, что в повреждении печени могут играть определенную роль и другие генетические и экологические факторы.
б) Морфология. Дефицит альфа-1-антитрипсина характеризуется наличием в цитоплазме гепатоцитов округлых или овальных зернистых включений, которые при рутинном окрашивании гематоксилином и эозином выглядят ацидофильными и нечетко отграниченными от окружающей цитоплазмы. Включения всегда PAS-положительные и устойчивые к действию диастазы. Такие же включения, но меньшего размера определяются у лиц с генотипами PiMZ и PiSZ.
По неизвестным причинам большинство включений расположены в ге-патоцитах, окружающих портальные тракты. Кроме того, количество гепатоцитов с включениями у пациентов с поражением печени не коррелирует с тяжестью патологии. Патологические изменения у гомозигот с генотипом PiZZ варьируют от гепатита новорожденных с холестазом и фиброзом или без них до цирроза печени, развивающегося в детском возрасте, или медленно прогрессирующего хронического гепатита или цирроза печени, которые проявляются в более старшем возрасте.
Почти во всех наблюдениях основным отличительным признаком поражения печени является наличие PAS-положительных включений. Редко присутствуют жировые изменения гепатоцитов и тельца Маллори. Диагностически значимые включения альфа-1-антитрипсина у младенцев могут отсутствовать, на вероятный дефицит ai-антитрипсина может указывать жировой гепатоз.
в) Клинические признаки. У 10-20% новорожденных с дефицитом агантитрипсина развивается гепатит с холестатической желтухой. У подростков симптомы заболевания могут ассоциироваться с гепатитом или циррозом печени. Гепатит может постепенно самопроизвольно купироваться и заканчиваться полным выздоровлением, а может становиться хроническим и прогрессировать до цирроза печени. Заболевание может не манифестировать клинически вплоть до развития цирроза печени в более позднем возрасте.
У взрослых с генотипом PiZZ в 2-3% случаев наблюдается ГЦК, обычно в сочетании с циррозом печени. Основным методом лечения при тяжелом поражении печени является ортотопическая трансплантация. У пациентов с поражением легких самым важным моментом лечения является отказ от курения, т.к. оно усиливает развитие эмфиземы и деструкцию легочной ткани на фоне дефицита ai-антитрипсина.
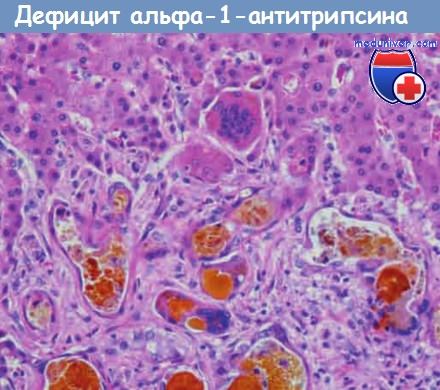
Признаки тяжелого холестаза.
- Вернуться в оглавление раздела "Патофизиология"
Оглавление темы "Патогенез заболеваний печени":- Морфология хронического вирусного гепатита
- Механизм развития (патогенез) молниеносного гепатита
- Механизм развития (патогенез) абсцесса печени
- Механизм развития (патогенез) аутоиммунного гепатита
- Механизм развития (патогенез) повреждения печени лекарствами
- Механизм развития (патогенез) повреждения печени алкоголем
- Механизм развития (патогенез) жировой дистрофии печени
- Механизм развития (патогенез) гемохроматоза
- Механизм развития (патогенез) болезни Вильсона-Коновалова
- Механизм развития (патогенез) дефицита альфа-1-антитрипсина