
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) болезни Хантингтона
Болезнь Хантингтона — это аутосомно-доминантное заболевание, характеризующееся прогрессирующими двигательными нарушениями и деменцией на фоне дегенерации стриарных нейронов. Типичной формой гиперкинеза является хорея, которая может захватывать любую часть тела.
Позднее присоединяются паркинсонический синдром с брадикинезией и ригидностью. Заболевание неуклонно прогрессирует, его продолжительность от начала симптомов до летального исхода составляет в среднем 15 лет.
а) Молекулярная генетика. Болезнь Хантингтона — это прототип заболеваний, ассоциированных с увеличением количества повторов триплетов нуклеотидов, кодирующих глутамин. Ген HD, расположенный на хромосоме 4р16.3, кодирует белок гентингтин массой 348 кДа. Первый экзон гена HD содержит ряд повторов кодона CAG, которые кодируют полиглутаминовый участок около N-конца аминокислотной цепочки белка.
В нормальном гене HD присутствует от 6 до 35 копий этого кодона. Заболевание развивается, если количество копий кодона превышает указанное число. Существует обратно пропорциональная связь между количеством повторов и возрастом манифестации заболевания, т.е. более длинная область повторов коррелирует с более ранним началом болезни. Поскольку эффекты этих повторов регулируют другие факторы, количество повторов не единственный предиктор возраста манифестации болезни.
Увеличение количества повторов происходит в процессе сперматогенеза, т.е. передача от отца генетического материала с мутацией потомству приводит к ранним проявлениям заболевания в следующем поколении — феномен антиципации. Новые мутации происходят редко. В большом количестве случаев спорадических форм заболевания может отсутствовать наследование по родительской линии, т.к. родители либо умерли до манифестации заболевания, либо в геноме у отца без манифестации заболевания присутствует небольшое количество повторов, которые усиливаются в процессе сперматогенеза.
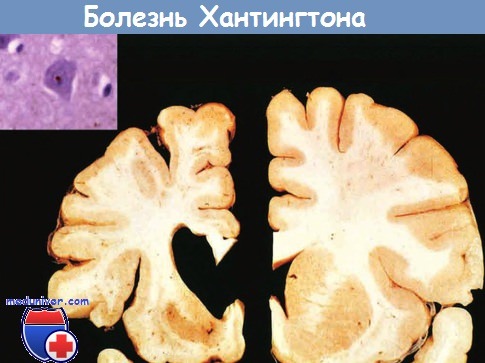
Слева — здоровое полушарие, справа — пораженное,
заметны выраженная атрофия стриатума и расширение просвета бокового желудочка.
Врезка: окрашивание внутриядерных включений при иммуногистохимическом исследовании.
б) Морфология. При макроскопическом исследовании обращают на себя внимание малые размеры головного мозга и ярко выраженная атрофия хвостатого ядра и в меньшей степени скорлупы (на ранних стадиях), атрофия бледного шара может развиваться вторично. Также наблюдается расширение боковых и третьего желудочков. Атрофия нередко присутствует и в лобных долях, реже — в теменных, иногда процесс охватывает всю кору.
При микроскопическом исследовании определяется деструкция стриарных нейронов, наиболее выраженная в хвостатом ядре, особенно в хвосте и вблизи просвета желудочка. Поражение скорлупы присоединяется на более поздней стадии заболевания. Патоморфологические изменения нарастают в хвостатом ядре в медиально-латеральном направлении, а в скорлупе — в дорсально-вентральном направлении. Наиболее сохранной частью стриатума является прилегающее ядро.
Наблюдается поражение как крупных, так и мелких нейронов, но сначала страдают мелкие нейроны. Шиповатые нейроны среднего размера, нейротрансмиттером в которых является y-аминомасляная кислота, а также энкефалин, динорфин и субстанция Р поражаются в наибольшей степени. Относительно интактными остаются две популяции нейронов: диафораза-положительные клетки, содержащие синтазу оксида азота, и крупные холинэстеразаположительные нейроны. Обе популяции выполняют функцию местных интернейронов.
Также наблюдается фибриллярный глиоз, который в данном случае более выражен, чем обычная реакция на гибель нейронов. Существует прямая связь между степенью дегенерации стриатума и выраженностью симптомов. Агрегаты, содержащие гентингтин, могут выявляться в нейронах стриатума и коры.
в) Патогенез. Гибель стриарных шиповатых нейронов приводит к нарушению функционирования системы базальных ядер, которая модулирует двигательные импульсы. В норме эти нейроны выступают в качестве демпферов активности двигательной коры, поэтому в результате их дегенерации при болезни Хантингтона развиваются гиперкинезы, чаще всего хореоатетоз. Присоединяющиеся когнитивные нарушения, вероятно, обусловлены деструкцией нейронов в коре больших полушарий.
Биологическая функция белка гентингтина пока неизвестна. Есть данные (но их немного), что заболевание развивается вследствие гаплоидной недостаточности, связанной с мутантным аллелем. Скорее всего, токсические свойства белка обусловлены дополнительными повторами в полиглутаминовом участке. Эти повторы вызывают агрегацию молекул белка и образование внутриядерных включений, которые, однако, не повреждают клетки непосредственно.
При болезни Хантингтона обнаружено нарушение регуляции транскрипции генов, поскольку мутантные формы гентингтина связывают важные регуляторы транскрипции, в т.ч. Sp1 и СВР (белок, связывающий цАМФ). Цепочка событий, ассоциированных с секвестрацией этих факторов транскрипции, предположительно заключается в снижении экспрессии PGC-1a, которая, в свою очередь, является фактором транскрипции, участвующим в митохондриальном биогенезе и механизмах защиты от окислительного повреждения.
г) Клинические признаки. Заболевание обычно манифестирует в возрасте 30-50 лет. Время появления симптомов зависит от длины участка повторов кодона CAG в гене HD. Двигательные нарушения обычно опережают когнитивный дефицит. Гиперкинезы при болезни Хантингтона хорееподобные с широкоамплитудными подергиваниями всех частей тела, характерны «скручивающие» движения конечностей. Ранние признаки утраты высших психических функций — нарушения памяти и аффективные расстройства, которые прогрессируют до глубокой деменции.
Среди лиц с болезнью Хантингтона повышен риск суицидов, однако ведущей причиной смерти является присоединение интеркуррентной инфекции. Поскольку скрининг позволяет выявить специфические для болезни Хантингтона мутации и очень тяжелое течение самого заболевания, оно часто становится объектом дискуссий об этичности генетической диагностики.
- Вернуться в оглавление раздела "Патофизиология"
Оглавление темы "Патогенез нервных болезней":- Механизм развития (патогенез) болезни Альцгеймера
- Механизм развития (патогенез) фронтотемпоральной деменции
- Механизм развития (патогенез) болезни Пика
- Механизм развития (патогенез) прогрессирующего надъядерного паралича
- Механизм развития (патогенез) кортикобазальной дегенерации
- Механизм развития (патогенез) сосудистой деменции
- Механизм развития (патогенез) болезни Паркинсона
- Механизм развития (патогенез) деменции с тельцами Леви
- Механизм развития (патогенез) множественной системной атрофии
- Механизм развития (патогенез) болезни Хантингтона