
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) болезни Паркинсона
Поражения этих отделов мозга часто проявляются такими двигательными расстройствами, как ригидность, нарушения позы и хорея. В целом эти поражения можно разделить на манифестирующие нарушением либо произвольных либо непроизвольных движений.
Важнейшую роль в положительной и отрицательной регуляции синаптических путей, которые обеспечивают обратную связь от таламуса к двигательной коре, играют базальные ядра, в первую очередь нигростриарный путь. Наиболее важными заболеваниями в данной группе являются паркинсонизм и болезнь Хантингтона.
Паркинсонизм — это клинический синдром, характеризующийся гипомимией, застывшей позой, брадикинезией, семенящей походкой, ригидностью и тремором с мелкими движениями пальцев рук, напоминающими катание шариков (тремор покоя по типу «скатывания пилюль»).
Этот вид двигательных расстройств наблюдается при различных патологических состояниях, общим для которых является поражение нигростриарной допаминергической системы. Паркинсонизм может быть лекарственно-индуцированным (на фоне приема агонистов допамина) и результатом отравления токсинами. К основным заболеваниям, при которых повреждается нигростриарная система, относятся:
- болезнь Паркинсона;
- множественная системная атрофия, обычно проявляющаяся паркинсонизмом и другими симптомами;
- постэнцефалитный паркинсонизм (осложнение пандемии гриппа в 1918 г.);
- прогрессирующий надъядерный паралич с кортикобазальной дегенерацией, при которых развиваются когнитивные нарушения.
Диагноз «болезнь Паркинсона» ставят при прогрессирующих признаках паркинсонизма (тремор, ригидность и брадикинезия), регрессирующих после терапии L-допой, и отсутствии токсических или иных причин. Существуют семейные формы болезни Паркинсона с аутосомно-доминантным и аутосомно-рецессивным типами наследования. Семейные формы встречаются реже, но их изучение позволило продвинуться в понимании патогенеза этого заболевания.
а) Морфология. Типичным макроскопическим признаком является депигментация черной субстанции и голубоватого пятна. При микроскопическом исследовании определяются уменьшение количества пигментных катехоламинергических нейронов в этих структурах и глиоз. В сохранных нейронах могут обнаруживаться тельца Леви.
Они представляют собой единичные или множественные эозинофильные цитоплазматические включения округлой или удлиненной формы, которые обычно имеют плотный центр, окруженный бледным ободком. При ультраструктурном анализе определяется, что тельца Леви представлены тонкими филаментами, плотно упакованными в центре и рыхло расположенными на периферии. Филаменты состоят из α-синуклеина. Тельца Леви могут быть обнаружены и в холинергических нейронах базального ядра Мейнерта, в котором количество нейронов снижается (особенно у пациентов с психическими нарушениями) так же, как и в стволовых ядрах, включая голубоватое пятно и дорсальное ядро блуждающего нерва.
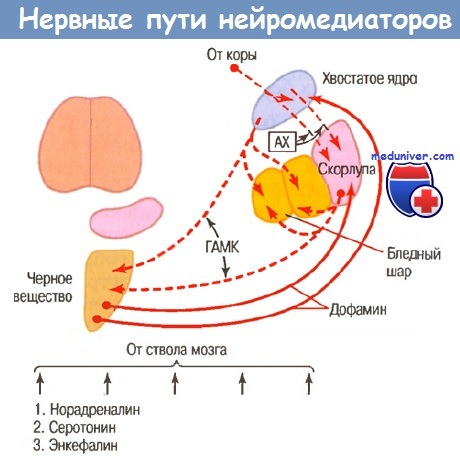
AX - ацетилхолин; ГАМК - гамма- аминомасляная кислота
б) Молекулярная генетика. Благодаря анализу групп сцепления обнаружено более десятка локусов генов, которые участвуют в развитии болезни Паркинсона, что указывает на сложный механизм развития болезни. Один из этих каузальных генов, обусловливающий аутосомно-доминантный тип болезни Паркинсона, кодирует α-синуклеин — широко распространенный в нервной системе белок, связывающий липиды, который в норме участвует в функционировании синапсов и является главным компонентом телец Леви.
Мутации гена, кодирующего α-синуклеин, наблюдаются редко; как правило, это точечные мутации, которые заключаются в амплификации хромосомы 4q21, где находится данный ген. Развитие болезни Паркинсона вызвано изменениями количества копий гена, которые усиливают его эффект (сравните АРР при болезни Альцгеймера). Предполагается, что риск развития болезни Паркинсона обусловливают полиморфизмы промотора α-синуклеина, меняющие его экспрессию. Мутации гена, кодирующего LRRK2 (обогащенная лейциновыми повторами киназа 2), — более частая причина аутосомно-доминантного типа болезни Паркинсона, но обнаружены в некоторых наблюдениях спорадической болезни Паркинсона.
Отдельные патогенетически значимые мутации усиливают киназную активность LRRK2, что указывает на роль этого феномена в развитии болезни Паркинсона.
Ювенильный аутосомно-рецессивный тип болезни Паркинсона ассоциируется с мутациями гена паркипа, кодирующего лигазу Е3, которая в результате связывания с различными субстратами утрачивает свою функцию. Патоморфологические характеристики паркин-обусловленной, α-синуклеин-обусловленной и спорадической форм болезни Паркинсона похожи, за исключением того, что в первом случае тельца Леви, как правило, отсутствуют.
Другие аутосомно-рецессивные формы болезни Паркинсона являются результатом мутаций гена, кодирующего белок DJ-1 (участвует в регуляции окислительно-восстановительных реакций при стрессе), либо гена, кодирующего киназу PINK1 (считается, что она задействована в регуляции нормальной функции митохондрий).
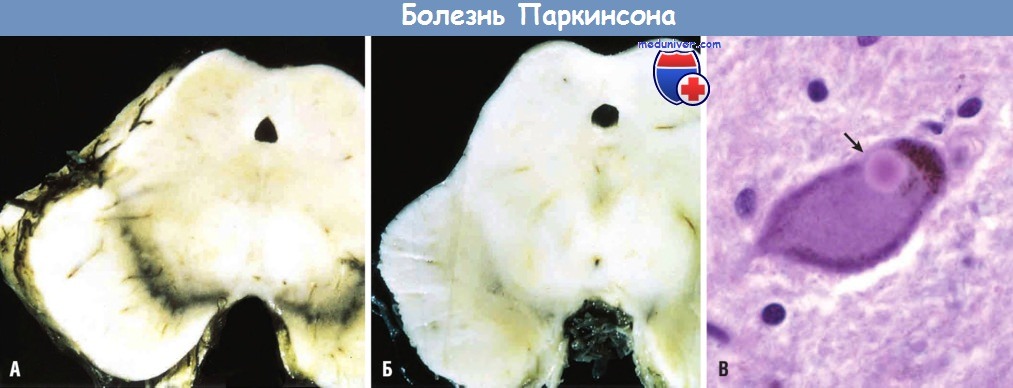
(А) Черная субстанция в норме.
(Б) Депигментированная черная субстанция при идиопатической болезни Паркинсона.
(В) Тельце Леви в нейроне черной субстанции, окрашенное в ярко-розовый цвет (стрелка).
в) Патогенез. С помощью генетических и биохимических исследований не удалось определить единый патогенетический механизм, поэтому на сегодняшний день есть несколько гипотез: (1) нарушение агрегации а-синуклеина в ответ на стресс; (2) дефект функции протеосом в связи с утратой функции паркина, убиквитин-лигазы Е3; (3) функциональные изменения в митохондриях, обусловленные отсутствием DJ-1 и PINK1.
Интересно, что на роль митохондриальной дисфункции указывают и другие факты. Например, содержание митохондриального комплекса I (компонента каскада окислительного фосфорилирования) в головном мозге пациентов со спорадической формой болезни Паркинсона снижено. Кроме этого, удалось воспроизвести болезнь Паркинсона в эксперименте, используя ингибиторы митохондрий.
Отростки допаминергических нейронов черной субстанции проецируются на стриатум, поэтому их дегенерация при болезни Паркинсона приводит к снижению уровня допамина в стриатуме. Выраженность акинетико-ригидного синдрома пропорциональна степени дефицита допамина, который можно, по меньшей мере частично, компенсировать путем заместительной терапии L-допой (промежуточный продукт цепочки синтеза допамина).
Однако такая терапия не может повернуть вспять уже происшедшие морфологические изменения и не останавливает развитие заболевания; более того, по мере прогрессирования болезни Паркинсона эффективность лечения снижается, и неврологические нарушения все труднее поддаются коррекции. Острый паркинсоноподобный синдром с деструкцией нейронов в черной субстанции развивается под действием МРТР (1-метил-4-фенил-1,2,3,6-тетрагидропиридин) — побочного вещества, которое присутствует в запрещенных к использованию аналогах психоактивного вещества миперидина.
Введение МРТР лабораторным животным позволило создать экспериментальную модель, успешно используемую при изучении методов лечения болезни Паркинсона, включая трансплантацию. Эпидемиологические исследования выявили, что пестициды являются фактором риска развития болезни Паркинсона, в то время как кофеин и никотин обладают протективным действием.
г) Клинические признаки. Помимо классических симптомов паркинсонизма характерны вегетативные нарушения, а в ряде случаев — когнитивные расстройства. Болезнь Паркинсона иногда сочетается с деменцией на своей ранней или поздней стадии. Применение L-допы в качестве симптоматического лечения, как правило, дает очень хороший эффект, но препарат не останавливает прогрессирование заболевания. Со временем эффективность препарата снижается и присоединяются двигательные флуктуации.
Поскольку биохимические нарушения при болезни Паркинсона давно и хорошо известны, одним из первых направлений исследований были нейротрансплантация и генная терапия. Широко применяют нейрохирургический метод лечения болезни Паркинсона, заключающийся в имплантации постоянных стимулирующих электродов на одном уровне экстрапирамидной системы для компенсации утраченной функции нигростриарной системы.
Видео этиология, патогенез болезни Паркинсона
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) деменции с тельцами Леви"
Оглавление темы "Патогенез нервных болезней":- Механизм развития (патогенез) болезни Альцгеймера
- Механизм развития (патогенез) фронтотемпоральной деменции
- Механизм развития (патогенез) болезни Пика
- Механизм развития (патогенез) прогрессирующего надъядерного паралича
- Механизм развития (патогенез) кортикобазальной дегенерации
- Механизм развития (патогенез) сосудистой деменции
- Механизм развития (патогенез) болезни Паркинсона
- Механизм развития (патогенез) деменции с тельцами Леви
- Механизм развития (патогенез) множественной системной атрофии
- Механизм развития (патогенез) болезни Хантингтона