
MedicalPlanet
Контактные данные:
admin@medicalplanet.su
2009-2023
Механизм развития (патогенез) болезни Альцгеймера
Главным представителем нейродегенеративных заболеваний с пораженим коры больших полушарий является болезнь Альцгеймера, которая проявляется деменцией — прогрессирующей утратой когнитивных функций вне зависимости от напряженности внимания. Существует несколько видов деменции: фронтотемпоральная деменция, сосудистая деменция, деменция с тельцами Леви, болезнь Крейтцфельда-Якоба и нейросифилис. При этих заболеваниях также поражаются субкортикальные структуры, но многие симптомы связаны с изменениями в коре больших полушарий. Независимо от этиологии деменция не является нормальным процессом старения, а всегда служит проявлением патологии.
Болезнь Альцгеймера — самая частая причина деменции в пожилом возрасте. Заболевание проявляется постепенными нарушениями интеллекта и расстройствами в эмоциональной сфере и поведении. По мере прогрессирования поражения коры присоединяются дезориентация, мнестические нарушения и афазия. В течение 5-10 лет пациент становится инвалидом, недоступным для контакта и лишенным способности к самостоятельному передвижению.
Симптомы редко возникают до 50 лет, однако частота заболевания с возрастом увеличивается в 2 раза каждые 5 лет: в возрастной группе от 60 до 64 лет частота составляет 1 %, в возрасте от 85 до 89 лет — 40% и выше. Прогрессирующая частота заболевания в связи с возрастом создала множество медицинских, социальных и экономических проблем в странах со стареющим населением. Большинство случаев болезни являются спорадическими, на долю семейной формы приходится лишь 5-10%. Тем не менее изучение именно семейного заболевания пролило свет на патогенез спорадической формы.
Хотя морфологическое исследование ткани мозга по-прежнему необходимо для постановки достоверного диагноза, современная клинико-рентгенологическая диагностика позволяет определить наличие болезни Альцгеймера в 80-90% случаев.
а) Морфология. При аутопсийном исследовании головного мозга видна атрофия коры различной степени выраженности с расширением борозд. Эти изменения особенно заметны в лобной, височной и теменной долях. Из-за выраженной атрофии происходит расширение желудочковой системы (гидроцефалия ex vacuo), поскольку объем мозговой ткани уменьшается. Поражение медиальных отделов височной доли, в т.ч. гиппокампа, энторинальной коры и миндалевидного комплекса, происходит раньше всего, поэтому на поздних стадиях болезни атрофические изменения этих структур выражены особенно сильно.
Главной характеристикой болезни Альцгеймера, позволяющей поставить гистологический диагноз при микроскопическом исследовании, являются сенильные бляшки и сети нейрофибрилл. Наблюдаются прогрессирующая и впоследствии массовая гибель нейронов и реактивный глиоз в местах образования сенильных бляшек и сетей нейрофибрилл.
Сенильные (нейритные) бляшки представляют собой фокальные сферические скопления расширенных и извитых отростков нейронов (дистрофически измененных отростков), расположенных обычно вокруг амилоидного центра, который может быть окружен светлым ободком. Диаметр сенильных бляшек варьирует от 20 до 200 мкм, по периферии располагаются клетки микроглии и реактивные астроциты. Сенильные бляшки обнаруживаются в гиппокампе, миндалевидном комплексе и неокортексе, однако поражение первичной двигательной и соматосенсорной коры выражено в меньшей степени (что также справедливо и для сетей нейрофибрилл).
Амилоидный центр, который можно выявить при окрашивании конго красным, содержит патологический белок. Главный компонент амилоидного центра сенильных бляшек — белок Ар, производное более крупной молекулы — АРР. Аминокислотные последовательности двух основных разновидностей белка Aβ, обозначаемых Aβ40 и Aβ42, имеют одинаковый N-конец и отличающийся на 2 аминокислотных остатка С-конец. Другие белки, присутствующие в сенильных бляшках, малочисленны, к ним относятся компоненты системы комплемента, провоспалительные цитокины, а1-антихимотрипсин и аполипопротеины.

Атрофия коры больше выражена в правом полушарии,
где удален лептоменинкс.
В ряде случаев происходит отложение белка Аβ со всеми тинкториальными характеристиками амилоида в отсутствие окружающей нейрональной реакции. Эти образования, называемые диффузными сенильными бляшками, обнаруживаются в поверхностных слоях коры больших полушарий, а также в базальных ядрах и коре мозжечка. Диффузное поражение является маркером ранней стадии формирования зрелых сенильных бляшек. Это заключение было сделано при исследовании мозга лиц с трисомией по 21-й хромосоме (синдромом Дауна). Для лиц с синдромом Дауна характерно более раннее начало болезни Альцгеймера.
В некоторых участках мозга (коре мозжечка и стриатуме) диффузные сенильные бляшки сами по себе или наряду с другими изменениями отражают пик развития болезни Альцгеймера. Сенильные бляшки содержат и Aβ40, и Aβ42, а диффузные сенильные бляшки состоят преимущественно из Aβ42.
Сети нейрофибрилл — это пучки филаментов в цитоплазме нейронов, смещающие или окружающие ядро. В пирамидных клетках пучки филаментов внешне часто напоминают рисунок языков пламени; в клетках более округлой формы переплетения волокон вокруг ядра формируют шаровидные сети нейрофибрилл. Эти сети имеют базофильную окраску при окрашивании гематоксилином и эозином и очень хорошо импрегнируются серебром. Сети нейрофибрилл обнаруживаются, как правило, в корковых нейронах, особенно в энторинальной коре, но могут иметь и другую локализацию.
Сети нейрофибрилл встречаются, например, в пирамидных клетках гиппокампа, миндалевидном комплексе, базальных отделах лобных долей и ядрах шва. Сети нейрофибрилл нерастворимы, устойчивы к элиминации и длительно сохраняются после гибели пораженного нейрона в качестве «могильных камней». При ультраструктурном исследовании видно, что сети нейрофибрилл состоят преимущественно из парных спиралевидных филаментов, расположенных вдоль нескольких прямых филаментов, имеющих сходный состав. Основным компонентом парных спиралевидных филаментов являются аномальные гиперфосфорилированные формы тау-белка, который присутствует в микротрубочках аксонов и способствует их соединению.
Другими компонентами являются МАР2 (белок, связанный с системой микротрубочек) и убиквитин. Парные спиралевидные филаменты обнаруживаются также в дистрофически измененных отростках нейронов, которые образуют наружные слои сенильных бляшек, и в аксонах, проходящих через пораженное серое вещество (нити нейропиля). Сети нейрофибрилл выявляются при других заболеваниях, т.е. не специфичны для болезни Альцгеймера.
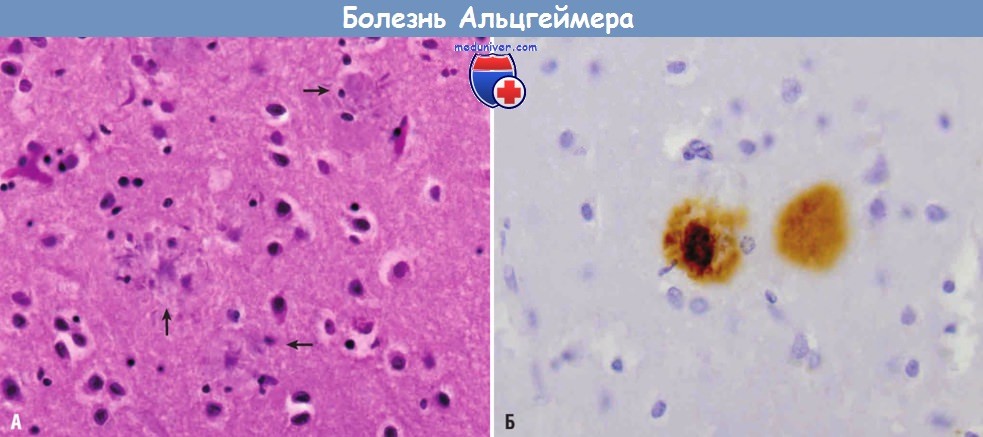
(А) Сенильные бляшки с дистрофическими отростками нейронов вокруг амилоидного центра (стрелки).
(Б) В центре сенильной бляшки и окружающем нейропиле с помощью иммуногистохимического исследования выявляется белок Aβ.
Для болезни Альцгеймера характерны и другие патоморфологические изменения. Церебральная амилоидная ангиопатия — неизменный спутник болезни Альцгеймера, но встречается и у лиц, не страдающих болезнью Альцгеймера. При церебральной амилоидной ангиопатии в сосудах откладывается преимущественно Aβ40. Гранулярно-вакуольная дегенерация представляет собой образование мелких (5 мкм в диаметре) светлых вакуолей в цитоплазме нейронов, каждая их которых содержит аргирофильные гранулы.
Вакуоли могут возникать при нормальном процессе старения, но при болезни Альцгеймера такие изменения выявляются преимущественно в гиппокампе и обонятельных луковицах. Тельца Хирано, очень характерные для болезни Альцгеймера, представляют собой удлиненные стекловидные эозинофильные структуры, состоящие из рядов, построенных из паракристаллиновых филаментов, основным компонентом которых является актин. Тельца Хирано обнаруживаются в основном в пирамидных нейронах гиппокампа.
Поскольку изредка сенильные бляшки и сети нейрофибрилл могут быть у лиц, не страдающих деменцией, окончательный диагноз ставят на основании клинических и патоморфологических данных. Прогрессирование болезни идет непрерывно. Патоморфологические изменения (появление сенильных бляшек, сетей нейрофибрилл, гибель нейронов и глиальная реакция) раньше всего появляются в энторинальной коре, затем распространяются через гиппокампальную формацию на мезокортекс и, наконец, достигают неокортекса. Сенильные бляшки оценивают в каждом участке коры (отсутствуют, мало, умеренное количество, много), а сети нейрофибрилл описывают по распространенности в головном мозге.
По этим характеристикам в сочетании с критериями NIA-Reagan устанавливают причастность патоморфологических изменений, типичных для болезни Альцгеймера, к развитию деменции у данного пациента.
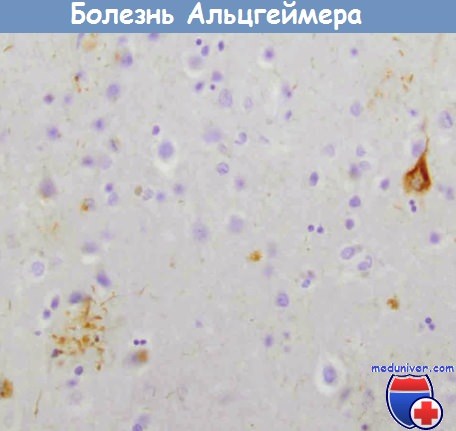
Сеть нейрофибрилл (вверхуслева) и нейриты вокруг сенильной бляшки (внизу справа),
содержащей тау-белок (иммуногистохимическое исследование).
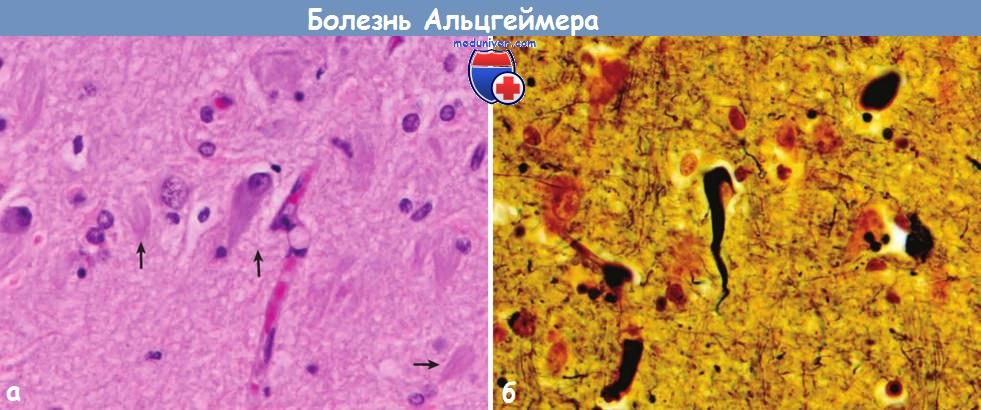
(А) Внутри нейронов видны сети нейрофибрилл, которые присутствуют и во внеклеточном пространстве (стрелки).
(Б) При импрегнации серебром видны сети нейрофибрилл в цитоплазме нейронов.
б) Молекулярная генетика и патогенез. В основе развития болезни Альцгеймера лежит отложение белков Aβ, которые образуются в результате процессинга АРР. АРР — это поверхностный белок, содержащий один трансмембранный домен. АРР может выполнять функцию рецептора, хотя лиганды этого белка до сих пор не обнаружены. Фрагмент Aβ АРР простирается от внеклеточного пространства до трансмембранного домена. Процессинг АРР начинается с расщепления внеклеточной части домена, затем расщепляется внутримембранная часть домена. Существует два пути, которые различаются по начальной стадии протеолиза. Если первое расщепление происходит в сайте связывания а-секретазы на участке последовательности, соответствующей Aβ, то белок Aβ не образуется (неамилоидогенный путь).
Такой процессинг АРР обычно происходит на поверхности клетки, где различные ферменты с а-секретазной активностью расщепляют поверхностные белки. Поверхностный АРР также может быть подвергнут эндоцитозу, тогда его расщепление происходит с помощью b-секретазы, которая разрывает цепочку аминокислот у N-конца Aβ (амилоидогенный путь). После расщепления АРР в одном из сайтов, у-секретаза осуществляет расщепление внутримембранной части домена. Если первый этап был выполнен а-секретазой, то образуется растворимый фрагмент, если b-секретазой — продуктом расщепления является белок Aβ. Различия в длине белков (Aβ40 vs Aβ42) обусловлены вариабельностью места разрыва аминокислотной цепочки при ее расщеплении у-секретазой. у-Секретазный комплекс, содержащий пресенилин, никастрин, pen-2 и aph-1, также участвует в процессинге сигнальной молекулы Notch, которая наряду с множеством других мембранных белков определяет судьбу клетки.
Белок Aβ имеет очень большую склонность образовывать агрегаты — сначала олигомеры (токсичные для нейронов), затем крупные агрегаты и фибриллы.
Изучение семейных форм болезни Альцгеймера позволило получить доказательства ведущей роли белка Aβ в инициации патологических событий, приводящих к болезни Альцгеймера. Ген, кодирующий АРР, находится на 21-й хромосоме, трисомия по которой ответственна за синдром Дауна. Патоморфологические изменения, характерные для болезни Альцгеймера, обусловливают когнитивные нарушения у лиц с этим синдромом. Характерные для болезни Альцгеймера гистологические изменения появляются в возрасте 10-30 лет, а неврологический дефицит развивается на 20 лет позже. Похожий эффект «умножения генов» наблюдается при локальной дупликации 21-й хромосомы, которая приводит к расширению локуса АРР у некоторых пациентов с семейной формой болезни Альцгеймера.
Точечные мутации гена, кодирующего АРР, тоже могут приводить к появлению семейной формы болезни Альцгеймера. Сайты некоторых мутаций находятся вблизи сайтов связывания b-секретазы и у-секретазы, другие локализуются в пределах последовательности Aβ АРР и усиливают способность белка к агрегации. Два локуса, ответственные за развитие большинства случаев семейной формы болезни Альцгеймера с ранним началом, кодируют два пресенилина: PS1 на 14-й хромосоме и PS2 на 1-й хромосоме. Мутации в этих локусах приводят к появлению патологических функций, например усиленной выработки у-секретазой белка Aβ, особенно Aβ42. Таким образом, генетические данные доказывают ведущую роль белка Aβ в патогенезе болезни Альцгеймера.
Белки Aβ сразу образуют агрегаты, обладающие нейротоксичностью. Есть свидетельства, что мелкие агрегаты вызывают дисфункцию синапсов путем долговременного блокирования передачи синаптических сигналов и изменений свойств других мембран других клеток. Агрегаты очень плохо распадаются, но мономерный Aβ может быть лизирован протеазами. И мелкие, и крупные агрегаты вызывают воспалительную реакцию со стороны микроглии и астроцитов. Не исключено, что эта реакция способствует удалению агрегатов, однако в то же время индуцирует секрецию повреждающих медиаторов. Другими следствиями активации каскада воспалительных реакций являются изменения процесса фосфорилирования и окислительное повреждение нейронов.
Генный локус на 19-й хромосоме кодирует апоЕ, который влияет на риск развития болезни Альцгеймера. Из-за полиморфизма двух аминокислот существуют 3 аллеля белка апоЕ (е2, е3 и е4). Увеличенная порция аллеля е4 повышает риск болезни Альцгеймера и снижает возраст манифестации заболевания. Установлено, что среди всех пациентов с болезнью Альцгеймера носители этого аллеля составляют абсолютное большинство. Аллель е4 вызывает образование белка Aβ и отложение его агрегатов по неизвестному механизму. В целом этот аллель ответственен за 25% случаев спорадической формы болезни Альцгеймера. Вероятно, другие аллели тоже являются факторами риска, но со значительно меньшим влиянием в популяции. В выявлении этих, более слабых локусов может быть полезен внедряемый новый подход к геномному скринингу.
Известно, что сети нейрофибрилл содержат тау-белок, поэтому его роль в развитии болезни Альцгеймера представляет большой интерес. Tay-белок связан с системой микротрубочек аксонов. При образовании сетей нейрофибрилл на фоне болезни Альцгеймера этот белок перемещается в тела и дендриты нейронов, где происходит его гиперфосфорилирование и он утрачивает способность к связыванию с микротрубочками.
Считается, что главным триггером развития болезни Альцгеймера является образование патологической формы белка Aβ, а не влияние тау-белка, поскольку мутации гена, кодирующего белок Aβ, приводят к формированию сетей нейрофибрилл и развитию болезни Альцгеймера, а мутации гена МАРТ, кодирующего тау-белок, вызывают развитие одной из форм фронтотемпоральной деменции, но не накопление белка Aβ. Механизм повреждения нейронов сетями нейрофибрилл мало изучен.
Вопрос о морфологическом субстрате деменции у пациентов с болезнью Альцгеймера остается открытым, однако доказана тесная взаимосвязь широкой распространенности сенильных бляшек и сетей нейрофибрилл с грубыми когнитивными нарушениями, причем количество сетей нейрофибрилл сильнее коррелирует со степенью выраженности деменции, чем количество сенильных бляшек. К биохимическим маркерам, коррелирующим с выраженностью деменции, относятся дефицит холинацетилтрансферазы, иммунореактивность синаптофизина и распространенность отложений амилоида.
в) Клинические признаки. Прогрессирование болезни Альцгеймера — длительный и непрерывный процесс (длительность симптоматического течения часто превышает 10 лет). Начальными симптомами являются мнестические нарушения, по мере развертывания клинической картины заболевания присоединяются речевые расстройства, акалькулия, утрата приобретенного динамического праксиса. Для терминальной стадии болезни Альцгеймера характерны нарушения контроля функции тазовых органов по типу недержания, мутизм и невозможность самостоятельного передвижения. Часто наблюдаются интеркуррентные заболевания (прежде всего пневмония), которые, как правило, приводят к смерти пациента. Одним из важнейших направлений исследований болезни Альцгеймера является выявление специфических биомаркеров. В настоящее время используют позитронно-эмиссионную томографию с амилоидсвязывающим препаратом PiB.
Существует 2 пути процессинга: неамилоидогенный (расщепление с помощью β-секретазы и y-секретазы) и амилоидогенный (путь, который приводит к образованию агрегатов белка Аβ и амилоидных фибрилл).
Видео этиология, патогенез болезни Альцгеймера (деменции)
- Рекомендуем ознакомиться со следующей статьей "Механизм развития (патогенез) фронтотемпоральной деменции"
Оглавление темы "Патогенез нервных болезней":- Механизм развития (патогенез) болезни Альцгеймера
- Механизм развития (патогенез) фронтотемпоральной деменции
- Механизм развития (патогенез) болезни Пика
- Механизм развития (патогенез) прогрессирующего надъядерного паралича
- Механизм развития (патогенез) кортикобазальной дегенерации
- Механизм развития (патогенез) сосудистой деменции
- Механизм развития (патогенез) болезни Паркинсона
- Механизм развития (патогенез) деменции с тельцами Леви
- Механизм развития (патогенез) множественной системной атрофии
- Механизм развития (патогенез) болезни Хантингтона